
Sulfur Isotopic Analysis and Sulfur Source Study of Phosphorite-associated Sulfate from the Ediacaran Doushantuo Formation in Guizhou Province
-
摘要:
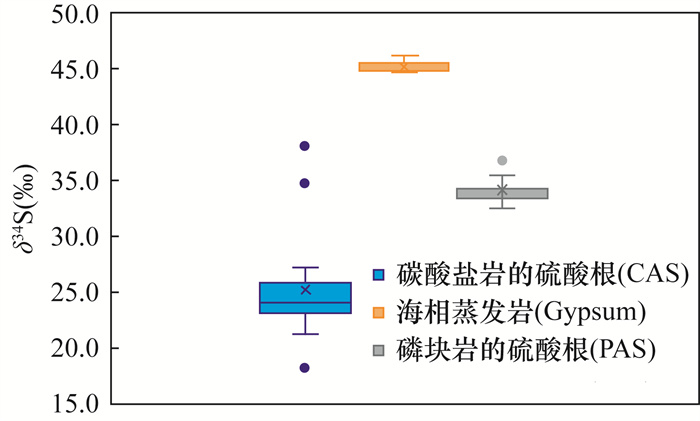
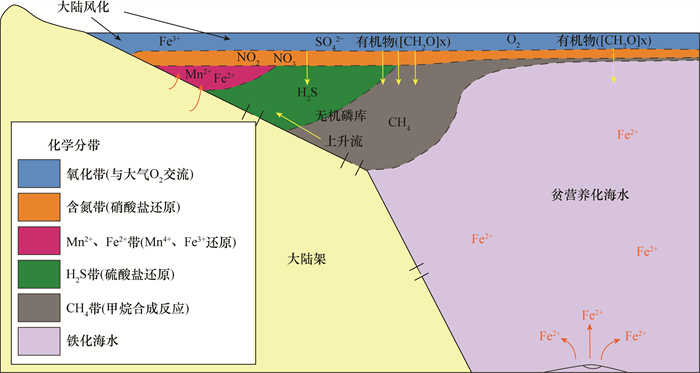
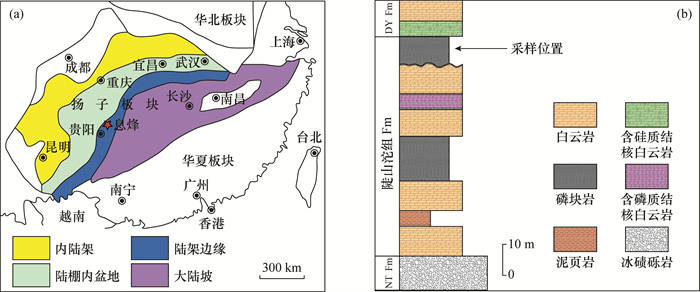
贵州地区埃迪卡拉系陡山沱组磷矿床沉积是新元古代晚期全球性成磷事件的典型代表,与气候突变及生命演化等存在密切联系。然而,目前对磷矿床沉积的研究仅局限于成磷机制和磷质来源等问题,而通过同位素地球化学指标研究该矿床成磷过程及与同期古海洋环境的关联研究较为薄弱。本文以贵州省息烽地区陡山沱组磷块岩为研究对象,在野外剖面观察和镜下岩石学特征研究基础上,利用元素分析仪-同位素质谱(EA-IRMS)连续流分析技术精确测得陡山沱组磷块岩的硫酸根硫同位素变化范围为32.7‰~36.9‰(n=32,平均值34.1‰),比同时期海水的硫同位素值低11‰,两者的差异表明磷块岩中的硫并不全部来自表层海水。理想化早期海洋(>520Ma)化学分带模型指示同时期海水中存在相对亏损34S的H2S带,结合磷块岩中磷质来源与上升流关系密切的认识,可以认为陡山沱组磷块岩的硫酸根硫同位素组成代表了表层海水和上升流的混合信号。
-
关键词:
- 元素分析仪-同位素质谱 /
- 磷块岩 /
- 磷块岩硫酸根硫同位素 /
- 陡山沱组 /
- 上升流 /
- H2S带
要点(1) 利用元素分析仪-同位素质谱法获得陡山沱组磷块岩的硫酸根硫同位素变化范围为32.7‰~36.9‰,与同时期海水硫组成比较,表明磷块岩中的硫并不全部来自表层海水。
(2) 早期海洋(>520Ma)化学分带模型揭示了磷块岩中的硫来自于H2S带。
(3) 磷块岩的硫酸根硫同位素值低于同时期海水,代表了上升流和表层海水的混合信号,或可成为指示海水硫酸根硫同位素的新指标。
HIGHLIGHTS(1) The sulfur isotopic composition of phosphorite-associated sulfate of the Doushantuo Formation ranged from 32.7‰ to 36.9‰ by elemental analyzer-isotope ratio mass spectrometry. Compared with the sulfur composition of seawater in the same period, it shows that the sulfur in the phosphate rock does not all come from the surface seawater.
(2) The early oceans (>520Ma) chemical zonation model indicated that sulfur of phosphorite came from the H2S zone.
(3) The sulfur isotopic composition of phosphorite-associated sulfate was lower than that of the contemporaneous seawater, representing a mixed signal of upwelling and surface seawater, which can be a new indicator of sulfur isotope of sulfate in seawater.
Abstract:BACKGROUNDPhosphate deposit of the Ediacaran Doushantuo Formation in Guizhou province is a typical representative of the global phosphorite formation event in the late Neoproterozoic, which is closely related to climate change and evolution of life. However, the current research on the deposition of phosphorus deposits is limited to the mechanism of phosphorus formation and the source of phosphorus, and research on the phosphorus formation process of this deposit and its correlation with the paleo-ocean environment of the same period by isotopic geochemical indicators is relatively weak.
OBJECTIVESIn order to determine the sulfur source of phosphorite-associated sulfate.
METHODSBased on the field section observation and the study of petrological characteristics under the microscope, elemental analyzer-isotope ratio mass spectrometry (EA-IRMS) was used to measure the sulfur isotopic composition of phosphorite-associated sulfate from the Ediacaran Doushantuo Formation. RESULTS: The sulfur isotopic composition of phosphorite-associated sulfate ranged from 32.7‰ to 36.9‰ (n=32, mean=34.1‰), which was 11‰ lower than that of the seawater of the same period, indicating that the phosphorite-associated sulfate was not all from the surface seawater.
CONCLUSIONSThe idealized early ocean (>520Ma) chemical zoning model indicates that there is a relatively 34S-depleted H2S zone in the seawater at the same time. Combined with the understanding that the source of phosphorus in the phosphorite is closely related to the upwelling, it can be considered that the sulfur isotopic composition of phosphorite-associated sulfate of the Doushantuo Formation represents the mixed signal of surface seawater and upwelling.
-
轻非水相液体(LNAPL)一般指不溶于水且比水轻的有机化合物(主要指原油、汽油、柴油、热媒油等碳氢化合物)。当LNAPL泄漏到地下时, 可产生垂向和横向迁移。LNAPL分布过程受土壤性质(如粒度和孔隙度)、非水相液体性质(如黏度)和LNAPL释放量等因素控制[1-3]。LNAPL在地下介质中残余导致的污染“拖尾&反弹”是污染修复的难点问题。监控自然衰减(Monitored Natural Attenuation, MNA)[4-5], 是国际上广为采用的低碳、绿色经济的污染场地修复技术。该技术出现于20世纪80年代,兴起于90年代, 至20世纪末已被广泛应用于污染场地修复。监控自然衰减技术与场地风险评价技术结合, 是污染场地“第二代”(“第一代”为工程修复)治理模式的核心, 被认为是当前解决LNAPL污染场地治理后污染物浓度发生“拖尾”和”反弹”现象的有效技术之一[6-7]。据不完全统计, 目前在美国非水相液体污染场地修复项目中, 监控自然衰减技术的应用率在70%~90%左右[4-8]。监控自然衰减技术可定义为[7]: 通过监测土壤和地下水中污染物及其相关指标, 揭示自然作用下场地污染物的时空动态过程及其机理, 并适时制定实施相应措施以引导自然作用, 控制土壤和地下水环境风险处于可接受水平以内。其内涵与中国当前生态文明建设“以自然恢复为主、人工修复为辅”的理念不谋而合。中国对监控自然衰减技术的研究开始较晚, 且以室内模拟试验为主, 目前实地应用案例比较罕见[8]。在技术规范方面, 直到2014年, 原国家环境保护部发布的《污染场地修复技术目录(第一批)》中才将自然衰减技术纳入地下水污染修复技术, 近些年关注度持续增高。
由定义可知, 监控自然衰减是以整个地下污染介质中污染物行为机制为研究对象。然而, 在2000年之前, 相关的研究主要侧重于地下水污染羽的自然衰减, 对污染物高度聚集的源区的自然衰减研究较少, 对存在非水相液体的源区自然衰减的研究更为罕见[9-11], 源区自然衰减的基本控制过程、影响因素、关键监控指标及转化速率等均不清楚[9]。在工程应用方面, 尽管美国国家环保署早在1997年的自然衰减文件中便指出源区控制是自然衰减修复项目的基本工作之一[12], 但受认知限制, 至今科学有效的源区控制仍未能很好地实现[9]。为了最大限度地控制源区污染风险, 现有的修复项目一般采用保守策略, 以主动修复技术(例如: 清除、围堵、强化生物修复、化学氧化、表面活性剂/助溶剂冲洗、热脱附等)移除污染源区的高浓度污染物, 再开展地下水污染羽自然衰减修复[9]。然而, 综合考虑修复有效性、费用经济性和风险控制合理性, 此类主动修复技术对源区污染的控制并不科学[9, 13-14]。如开挖之类的异位清除技术可移除非水相液体污染物, 但属于风险过度管控, 经费成本较高。相对经济的原位修复技术, 修复后期非水相液体污染物残余导致的污染“拖尾&反弹”问题几乎不可避免。在源区污染物行为机制不明的情形下, 关于何时适合进入地下水污染羽自然衰减修复的科学论证并不充分, 当前一般按最严格的策略制定主动修复目标值, 风险控制的合理性存疑、修复的费用偏高。如果污染场地的土壤渗透性较低, 则达标极为困难。因此, 为了解决上述技术和经济问题, 有必要加强对源区污染物行为机制的研究, 建立科学、有效的源区自然衰减监测评估方法, 系统认知源区-污染羽自然衰减, 以实现污染风险的合理控制[15-16]。
本文以LNAPL污染场地为对象, 以20世纪末监控自然衰减广泛应用为综述起点, 简要回顾了源区自然消除(Natural Source Zone Depletion, NSZD)技术的发展历程, 在阐述源区自然消除的定义内涵的基础上, 参考近些年源区研究成果, 重点介绍LNAPL污染源区垂向分区自然消除概念模型, 阐述关键控制因素。进而, 结合现有场地污染调查和监测技术现状, 归纳了NSZD方法的基本内容、一般程序和主要技术。最后, 提出了NSZD方法应用于污染场地修复需解决的关键科学技术问题及未来的研究发展趋势。
1. 源区自然消除(NSZD)技术的发展历程
1997年美国环境保护署OSWER指令指出[12]: 所有污染场地均需评估其源控制技术, 尤其对于采用监控自然衰减技术修复地下水污染羽的场地, 对污染源的评估更为重要。但其中并没有明确污染源区的内涵, 更没有明确提及开展源区自然衰减的具体内容。
2006年Paul Johnson等发表的系列论文[17-18], 概述了与源区的自然衰减有关的技术方法, 明确了源区自然衰减的概念内涵, 总结了一套分析污染物的衰减证据和评估源区自然衰减对烃类污染物浓度及组成的长期影响的方法, 并以某油田为例论证了方法的可行性和必要性。文中所述源区自然衰减(该文章缩写为Source Zone Natural Attenuation, SZNA)的概念与后续研究中广泛采用的源区自然消除(NSZD)的理念接近, 但SZNA的概念较为广义, 其本质上与污染羽内涵基本一致:包含对流-弥散、吸附-解吸、挥发、降解等多个过程, Paul Johnson等所提出的SZNA概念即为NSZD的雏形。
2009年美国州际技术与监管委员会(Interstate Technology & Regulatory Council, ITRC)出版的技术综述报告《Evaluating Natural Source Zone Depletion at Sites with LNAPL》, 进一步明确并提出了针对LNAPL的源区自然消除的内涵及量化评估方法[19], 即LNAPL挥发过程中的生物降解对污染物的消耗是源区自然消除的主要研究对象, 观测挥发过程中气体组成及其通量是量化评估核心内容, 此标志着NSZD的基本理论框架和评估技术体系构建方向的确立。
近十年来[20-31], 深入认知NSZD机制, 完善土壤-地下水一体化评估, 成为监控自然衰减研究的目标, 研究内容已由地下水污染羽自然衰减为主, 转变为源区LNAPL自然消耗研究为主, 发生了研究范式的转变(表 1)。
表 1 自然衰减的研究范式Table 1. Research paradigm of natural attenuation项目 地下水污染羽自然衰减(1990—2000年) 非水相液体污染物(LNAPL)污染源区自然消除(2000年至今) 管理重点 污染羽迁移多远 源区持续时间多长 主要污染物 溶解的苯系物 所有LNAPL成分 主要生物降解过程 电子受体介导的生物降解 产甲烷 包气带关键生物降解过程 LNAPL挥发蒸汽好氧生物降解 LNAPL厌氧生物降解(产甲烷)和好氧甲烷氧化 饱水带关键生物降解过程 溶解苯系物的厌氧生物降解 产甲烷脱气和冒泡, 厌氧生物降解LNAPL的研究 关键指标 微生物降解能力 NSZD速率 方法要点 上下游电子受体和降解产物的对比 二氧化碳通量; 包气带耗氧量梯度; 热通量 典型衰减速率 苯系物的半衰期为2~4年 NSZD的速率约100~1000加仑/英亩/年 2. 概念定义
2.1 非水相液体污染物源区定义
非水相液体污染物的源区可认为是可移动污染物和残余污染物存在的区域(图 1) [19]。其形成机制如下: 体积较小的LNAPL泄漏, 在到达地下水水面之前, 可能会被束缚在土壤孔隙中, 成为不可移动的残余相存在。体积相对较大的LNAPL泄漏, 可以到达地下水水面, 并在迁移路径上形成不可移动的残余相; 到达地下水水位处, LNAPL主要在饱水带界面累积, 并沿着地下水流向展布。在各种自然衰减机制(如吸附、挥发、溶解等)作用下, LNAPL的组成成分和化学性质均会随时间变化而改变。如果没有被去除, LNAPL污染“体”可能成为潜在长效污染源, 对邻近土壤、土壤气和地下水产生二次影响。此LNAPL污染“体”即为LNAPL“源区”。其中包气带中的源区部分称为“包气带或裸露源区”; 饱水带或地下水位以下的源区部分称为“饱水带或淹没源区”。
2.2 非水相液体污染物源区自然消除基本内涵
理论上, 若存在挥发、溶解、生物降解和吸附等自然衰减作用, 导致一定数量的化学物质以某种速度从源区自然流失, 则源区的非水相液体污染物必然显示正在消耗。因此, 采用质量平衡方法进行简单、定量评估, 即可论证源区自然消除的发生[9, 19]。由此可见, 从概念上, 源区自然消除仍属于自然衰减, 是自然衰减在源区发生的特例。然而, 其内涵与传统研究的地下水污染羽自然衰减存在较大区别。在传统认知的地下水迁移导致污染物衰减作用方面, 更强调气体迁移导致的污染物衰减。以往研究主要认为源区LNAPL的衰减主要由蒸汽挥发作用导致。而源区自然消除强调迁移过程中污染物的彻底消除, 源区LNAPL的衰减主要由生物降解作用导致。当然, 随着对机理过程的认知不断深入, 源区自然消除的内涵也会更加丰富[19, 26, 29]。
2.3 非水相液体污染物源区自然消除的机理过程
Lari等[29]总结了当前LANPL源区自然消除相关的21个物理和生物过程(表 2), 主要包括四类: ①控制流体传输的物理化学过程; ②LNAPL分配和成分变化过程; ③包气带和地下水中的NSZD过程; ④支持NSZD的基本微生物过程。
表 2 源区自然消除(NSZD)机制的机理过程Table 2. Mechanism and processes of NSZD机理过程 简要说明 机理过程 简要说明 1. 挥发 LNAPL化合物转化为气相 12. 饱水带源区自然衰减 饱水带的分配、非生物和生物降解 2. 溶解 LNAPL化合物转化为水相 13. 微生物迁移 微生物通过对流-弥散作用迁移 3. 毛细作用 压差导致的浸润液体在细管里升高和不浸润液体在细管里降低 14. 生物膜生长/脱落 附着在土壤颗粒上的微生物菌落的增厚和脱落 4. 多相流传输 各相流体的流动 15. 微生物营养物 基质或营养物质的微生物吸收活性 5. LNAPL残余 形成不可移动的LNAPL 16. 抑制因子 基质利用或生物量形成的限制因素 6. LNAPL成分的异质性 LNAPL成分的变化 17. 食物链 捕食者(如原生动物)和猎物(如细菌)的存在 7. 阻滞作用 取决于毛细作用对浸出和浸入过程的影响 18. 微生物差异 微生物在底物利用特性方面的差异 8. 介质的异质性 土壤性质的空间差异 19. 底物竞争 微生物同时消耗两种或两种以上底物 9. 饱和度变异 包气带含水饱和度的时空变化 20. 同化作用 微生物菌落生长初始滞后期 10.溶质传质 通过对流-弥散输送溶质 21. 气泡过程 生物降解使水相出现气泡 11.包气带源区自然衰减 包气带的分配、非生物和生物降解 2.4 非水相液体污染物源区自然消除的基本过程
源区自然消除的机理过程关联复杂, 对于实际应用来说, 逐一揭示每个过程并不现实。因此2009年, 美国ITRC综述报告[19]中对LNAPL源区自然消除及其基本过程进行了明确, 并对NSZD速率、机理过程及应用前景作了介绍。其认为: 当物理作用使LNAPL组分重新分配到水相或气相, 或者污染源相关组分发生生物降解时, 均视为LNAPL发生了NSZD过程。NSZD可视为污染场地LNAPL总量减少的综合过程, 包括LNAPL组分通过溶解于地下水和挥发至包气带等物理作用重新分配, 进而这些溶解或挥发的LNAPL组分被微生物降解消除。即, LNAPL溶解-饱水带中生物降解、LNAPL挥发-包气带中生物降解, 这两个过程是源区自然消除两个基本过程[19, 26, 29]。另外, LNAPL直接生物降解也是基本过程之一, 但该过程是否足够有效, 还有待论证。
2.4.1 非水相液体污染物溶解-饱水带中生物降解过程:饱水带自然消除
根据LNAPL组分的水溶性, 淹没在地下水位以下的LNAPL部分组分会被溶解到地下水中。溶解相成分通过地下水的运移离开源区, 并发生生物降解, 本文将该此过程简称为“饱水带自然消除”。实际上该过程基本可视为污染羽自然衰减, 即传统的监控自然衰减研究所关注的过程。近些年研究发现, 这个过程仅占LNAPL总质量损失的1%~10%。可见, 传统的监控自然衰减评估大大低估了污染物的消除量。图 2描述了地下水源区溶解和生物降解过程的基本要素[19]。
地下水溶解和生物降解过程的NSZD机制主要由以下因素控制: ①溶解度和有效溶解度; ②电子受体的可用性; ③地下水流场。从源区回收LNAPL可导致LNAPL降低至残余饱和度, 同时根据其溶解度, LNAPL在饱和区会持续溶解。
2.4.2 非水相液体污染物挥发-包气带中生物降解过程:包气带自然消除
包气带内LNAPL的烃类成分挥发进入土壤气体。然后, 碳氢化合物蒸汽通过扩散和平流在包气带土壤中迁移, 并发生生物降解, 本文将此过程简称为“包气带自然消除”。近些年研究认为, 该过程是源区自然消除的主要作用, 占LNAPL总质量损失的90%~99%, 此过程是当前源区自然消除研究的主要对象[19, 26, 29]。这个过程中, 扩散主要指挥发物从高浓度区转移到低浓度区。在地下, 蒸汽优先从地下污染源区向地表迁移。大多数自然条件下, 扩散通常是非饱和多孔介质中气相传输的主要机制, 是所含组分的气体扩散系数和土壤充气孔隙度的函数[32-36]。在有效孔隙率较高的土壤中(如含水量较低的砂土), 扩散和水汽迁移较快。平流主要指由压力梯度驱动的土体气体运动。土体气体从高压区流向低压区, 抽气井能引起包气带土壤中土体气体的平流运动。自然和人工气压梯度可引起土体气体的平流, 并导致蒸汽侵入到上层建筑内。夹薄层的地层(如, 中砂夹粉土)可对非饱和土壤的蒸汽传输产生显著影响[37-41]。
目前的文献中, 对污染物蒸汽迁移过程的生物降解研究较多。大量研究证实[25-27, 42-47], 生物降解是包气带蒸汽衰减的主要原因, 可大大缩短可生物降解组分的扩散距离和耗尽时间, 降低最大气体浓度, 特别是对于水溶性高的组分和亨利常数低的组分(例如苯)。从蒸汽入侵风险的角度来看, 生物降解是影响蒸汽迁移的关键因素, 以苯蒸汽为例, 不考虑生物降解的蒸汽迁移模型可能会高估蒸汽入侵风险500~1000倍。
2.4.3 LNAPL直接生物降解
关于LNAPL直接生物降解的文献相对较少。尽管通常认为, 源区污染物的生物降解会受到LNAPL-水的分配速率的限制[47-48], 但一些实验室研究表明, 溶解在溶剂中的目标组分矿化速率超过了分配速率, 指示着生物降解作用的存在[46-47]。有研究提出了各种以细菌促进LNAPL组分生物降解的机制[47-48], 但目前这方面的研究成果有待于更多野外现场验证。
3. 非水相液体污染物场地的源区自然消除概念模型
3.1 非水相液体污染物场地概念模型
源区中LNAPL的分布特征取决于场地污染的年限。按距泄露发生的时间, 可将污染场地划分为早期、中期和晚期三个阶段[22]。
早期阶段, LNAPL处于正在泄露或泄露刚结束, 源区由一个连续的、可移动的LNAPL污染体组成, LNAPL可扩展和迁移。随着LNAPL污染体的扩大, LNAPL的总损失增加。自然损失率开始接近LNAPL输入率, LNAPL污染体的扩张速度减慢。中期阶段, LNAPL输入和自然损耗几乎相等, LNAPL污染体变得基本稳定或收缩。现场调查表明, 许多历史遗留LNAPL泄露场地处于中期阶段。晚期阶段, 自然损耗已去除大部分LNAPL, 剩余污染物以残余LNAPL的形式存在。
3.2 非水相液体污染物源区自然消除概念模型
Suthersan等[45]在ITRC基础上, 指出源区自然消除效果的90%来源于包气带自然消除。Irianni-Renno等[47]在美国西部某石油类污染场地系统阐述了NSZD过程及微生物特征。Garg等[26]基于此提出了一维简化概念模型。该概念模型依据NSZD机制关键过程, 对应地下水饱和带和包气带, 将LNAPL污染体之上的NSZD区域划分为产甲烷、甲烷氧化和有氧传输三个区。Askarani等[31]将地下水饱和带LNAPL下方的厌氧区也归入NSZD, 在Garg等[26]模型的基础上, 将NSZD概念模型划分为厌氧、产甲烷、甲烷氧化和有氧传输四个区。本文综合以往的研究, 以水文地质结构和生物作用类型为分区依据, 对NSZD的四分区概念模型进行了优化, 并对各区的气体传输的关键过程、潜在控制因素以及主要氧化还原反应等进行阐述, 详述如下。
3.2.1 饱和带污染羽的产二氧化碳区
该区完全位于地下水位以下, 且LNAPL已溶解于污染羽, 污染物浓度相对较低, 氧气等常见可用电子受体受限。该区域一般为厌氧环境, 微生物主要依次以硝酸盐、铁、锰、硫酸盐为电子受体, 以碳氢化合物为电子供体和碳源, 发生硝酸盐还原、锰还原作用、铁还原、硫酸盐还原等反应, 产生二氧化碳。因此, 电子受体供给是该区最重要的控制因素, 氧化还原电位、pH等可作为影响因素。该区发生的氧化还原反应如下(碳氢化合物以C10H22表示, 下同):
$$ \mathrm{C}_{10} \mathrm{H}_{22}+62 / 5 \mathrm{NO}_3^{-}+62 / 5 \mathrm{H}^{+} \rightarrow \\ 10 \mathrm{CO}_2+31 / 5 \mathrm{~N}_2+86 / 5 \mathrm{H}_2 \mathrm{O} \\ \mathrm{C}_{10} \mathrm{H}_{22}+ 62 \mathrm{Fe}(\mathrm{OH})_{3(\mathrm{~s})}+124 \mathrm{H}^{+} \rightarrow \\ 10 \mathrm{CO}_2+62 \mathrm{Fe}^{2+}+166 \mathrm{H}_2 \mathrm{O} \\ \mathrm{C}_{10} \mathrm{H}_{22}+ 31 \mathrm{MnO}_{2(\mathrm{~s})}+62 \mathrm{H}^{+} \rightarrow \\ 10 \mathrm{CO}_2+31 \mathrm{Mn}^{2+}+42 \mathrm{H}_2 \mathrm{O} \\ \mathrm{C}_{10} \mathrm{H}_{22}+31 / 4 \mathrm{SO}_4^{2-}+31 / 2 \mathrm{H}^{+} \rightarrow \\ 10 \mathrm{CO}_2+31 / 4 \mathrm{H}_2 \mathrm{~S}+11 \mathrm{H}_2 \mathrm{O} $$ 3.2.2 饱和带-包气带底部的产甲烷区
饱和带发生显著的产甲烷作用, 导致甲烷和二氧化碳排放产生气泡, 这一过程也发生在包气带底部, 混合气体垂直向上迁移。当污染源中含较轻的燃料成分(如汽油)时, 通过厌氧输送区的气体也可能包括挥发性有机物。这个区域的潜在控制因素包括: 温度、pH、电子受体、挥发性碳氢化合物、营养物质、地下水位波动等。主要发生的氧化还原反应为:
$$\mathrm{C}_{10} \mathrm{H}_{22}+9 / 2 \mathrm{O}_2 \rightarrow 31 \mathrm{CH}_4+9 / 4 \mathrm{H}_2 \mathrm{O} $$ 3.2.3 包气带的甲烷氧化区
甲烷和二氧化碳通过包气带向上迁移, 至氧气达到一定浓度的区域发生氧化反应, 大部分或全部甲烷和挥发性有机物在该区被氧化为二氧化碳。目前, 蒸汽入侵研究已经认识到包气带中氧化反应的重要性, 以此解译包气带中VOCs的衰减。该区的潜在控制因素包括气体扩散系数、土壤水分、土壤类型、温度等。主要发生的氧化还原反应为:
$$ \mathrm{CH}_4+2 \mathrm{O}_2 \rightarrow \mathrm{CO}_2+2 \mathrm{H}_2 \mathrm{O} $$ 3.2.4 包气带上部近地表的土壤呼吸区
该区域内氧气向甲烷氧化区扩散, 产生的二氧化碳通过包气带向地表扩散。好氧传输区的厚度(或到甲烷氧化区的深度)取决于甲烷生成区释放的甲烷和挥发性有机物的通量, 及进入地下的氧气通量。当甲烷和挥发性有机物通量相对较低时, 好氧传输区可延伸至非饱和区, 在该区发生好氧降解。如果浅层包气带也受到污染(例如, 在泄露点附近), 则浅层的好氧生物降解可能会限制氧气扩散到甲烷氧化区的量。如果浅层消耗的氧气超过了产甲烷区生成的甲烷完全被氧化所需的氧气, 则可能发生甲烷外逸。该过程仅存在于浅层污染情景下, 并不普遍。该区的潜在控制因素包括气体扩散系数、昼夜影响、季节性影响及气象(如风、降雨、气压等环境)因素影响等。该区发生的氧化还原反应为(土壤有机碳以CmHn表示):
$$ \mathrm{C}_m \mathrm{H}_n+(m+n / 4) \mathrm{O}_2 \rightarrow m \mathrm{CO}_2+n / 2 \mathrm{H}_2 \mathrm{O} $$ 另外, 由于NSZD通常适用于非水相液体泄露中晚期阶段的场地, 因此VOCs的直接挥发可能是许多场地的次要过程。当包气带较薄的情况下, VOCs较易迁移至近地表发生如下好氧反应:
$$ \mathrm{C}_{10} \mathrm{H}_{22}+31 / 2 \mathrm{O}_2 \rightarrow 10 \mathrm{CO}_2+11 \mathrm{H}_2 \mathrm{O} $$ 4. 非水相液体污染物源区自然消除的研究内容和方法
由以上分析可知, 源区自然消除过程中, 微生物降解速率受土壤和地下水中的电子受体(如:氧、硝酸盐、三价铁、四价锰、硫酸盐、二氧化碳等)的类型和可用性等因素控制。结合现有污染场地调查修复的技术流程[19, 26, 29], 本文将源区自然消除的主要研究内容归纳为LNAPL源区-羽识别、定性判断和定量估算三个方面, 分别阐述如下。
4.1 非水相液体污染物源区-羽识别
该项工作主要在污染场地的调查阶段开展, 目的是通过水文地质和污染调查, 对场地的污染状况和LNAPL的污染源与污染羽的分布进行识别和概括, 主要内容包括: 构建LNAPL污染场地概念模型、模拟地下水污染羽流过程和刻画污染源区三部分。
(1) 初步构建LNAPL场地概念模型。通过污染调查和水文地质调查的手段, 明确识别LNAPL污染体、溶解相羽流、蒸汽羽流、潜在暴露途径和敏感点。概念模型需体现出LNAPL的污染主体(水平和垂直范围)、地下水溶解相羽流和包气带蒸汽羽流。并绘制LNAPL概念性场地模型的平面和剖面图。
(2) 模拟地下水污染羽流过程。主要观测地下水污染羽中污染物(组分和浓度)和水化学指标, 分析饱水带NSZD的过程和速率。基于概念模型, 建模推断或预测LNAPL溶解速率、LNAPL可溶组分浓度、背景组分浓度和质量变化以及敏感点潜在风险等诸多信息。
(3) 刻画污染源区。通过对LNAPL源区的性质和分布的精细刻画, 确定包气带和饱和带(毛细带和饱水带)中LNAPL的分布, 估算源区污染物总量。其中, 与挥发相关的过程产生的源区消除效果通过包气带LNAPL(暴露源区)来计算, 与溶解相关的过程产生的源区消除效果通过饱和带LNAPL(淹没区)来计算。
4.2 非水相液体污染物源区自然消除的定性判断
该项工作主要在调查数据分析和修复方案制定阶段开展, 其目的是通过水文地质和污染调查数据, 分析污染源区的LNAPL向地下水的溶解、源区地下水中污染物的生物降解、源区污染物向包气带的挥发、源区挥发至包气带中的污染物的生物降解等四个过程的相关指标的变化情况(表 3)。提供源区LNAPL向地下水和包气带迁移再分配, 以及微生物降解的定性证据, 判断源区自然消除过程是否有效发生。
表 3 NSZD的定性判断所需数据Table 3. Data required for qualitative evaluation of NSZDNSZD过程 数据 用途 源区LNAPL向下游地下水溶解 地下水监测井的静态水位 确定水力梯度和地下水流向(与浓度数据联合分析) 源区上下游地下水中溶解相污染物浓度 相较上游井, 下游井中溶解相碳氢化合物浓度的增加, 表明发生了溶解过程 渗透系数 估算饱和区污染物溶解和生物降解的损失率 源区溶解于地下水中的污染物生物降解 源区上下游地下水中作为反应物的溶解电子受体(如O2, NO3-, SO42-)和产物(如Fe2+, Mn2+) 相较上游井, 下游井中作为反应物的溶解电子受体(如O2, NO3-, SO42-)降低、产物(如Fe2+, Mn2+)增加, 表明发生了生物降解过程 源区上下游地下水中的溶解甲烷(CH4) 相较上游井, 下游井中的溶解甲烷(CH4)增加, 表明发生了产甲烷作用 源区污染物向包气带挥发 土壤垂向剖面土壤气中碳氢化合物浓度 随着与源区距离的增加, 土壤气体中碳氢化合物的浓度降低, 表明发生了挥发作用 土壤中总石油烃组成 土壤中碳氢化合物组成发生变化, 表明发生了NSZD过程, 改变化是质量损失过程的综合效应体现 源区挥发至包气带的污染物生物降解 土壤垂向剖面呼吸和相关土壤气浓度(O2, CO2, CH4) 在源区, 随着深度的增加, 土壤气O2减少、CO2增加或CH4浓度增加, 表明发生NSZD过程(CH4也可能来自自然界, 可通过对比背景区的碳稳定同位素来区分) 土壤中TPH的浓度随时间变化 需要大量样本, 较难长期监测 有效扩散系数: 实测或经验公式估算(土壤湿度、总孔隙度) 估算挥发和生物降解造成的损失率(氧气输送或甲烷产生率) 4.3 非水相液体污染物源区自然消除的定量评估
该项工作视数据丰富程度, 可贯穿于整个场地工作过程。其主要目的是在源区自然消除的定性评价基础上, 通过碳氢化合物的化学计量系数换算(表 4)、土壤气扩散模型以及地下水溶质运移模型计算, 对源区自然消除的速率进行定量评估。可分为饱和带和包气带两个单元分别估算。
表 4 碳氢化合物的化学计量系数表(以C10H22计)Table 4. Conversion coefficients of hydrocarbons (calculated by C10H22)微生物降解过程 电子受体/代谢产物 化学计量系数(Si) 好氧降解 O2 0.29kg-HC/kg-O2 硝酸盐还原 NO3- 0.19kg-HC/kg-NO3- 铁还原 Fe2+ 0.041kg-HC/kg-Fe2+ 硫酸盐还原 SO42- 0.19kg-HC/kg-SO42- 锰还原 Mn2+ 0.083kg-HC/kg-Mn2+ 产甲烷 CH4 1.1kg-HC/kg-CH4 4.3.1 饱和带源区自然消除定量评估
饱和带源区自然消除的定量评价与传统污染羽自然衰减的评价基本相同, 只是将目标含水层定义于污染源区。以碳氢化合物(HC)为例, 源区饱和带自然消除的通用计算公式如下:
$$ \begin{aligned} R_{\text {Sat }}=&\left(V Q_{\mathrm{HC}_{\text {Horizontal( out-in) }}}+\Delta Q_{\mathrm{HC}_{\text {Vertical( out-in) }}}\right)+\\ & Q_{\mathrm{H}_2 \mathrm{O}}\left[\varSigma\left(S_{\text {electron donor }} \Delta C_{\text {electron donor }{ }_{\text {up-down }}}\right)+\right.\\ &\left.\sum\left(S_{\text {byproduct }} \Delta C_{\text {byproduct }_{\text {down-up }}}\right)\right] \end{aligned} $$ (1) 式中:RSat为源区饱和带中HC损失速率(kg-HC/d); ΔQHCHorizontal为水平方向物理作用输出-输入HC速率(kg-HC/d); ΔQHCVertical为垂直方向物理作用输出-输入HC速率(kg-HC/d); QH2O为地下水流量(L/d); Sbyproduct为电子受体(代谢产物)消耗HC的化学计量系数[kg-HC/kg-电子受体(代谢产物)]; ΔCelectron donorup-down为上游与下游地下水中电子受体浓度之差[kg-电子受体(代谢产物)/L]。
按照一般监控自然衰减的理论方法, 需切断目标含水层垂直方向物理作用输出-输入量, 则ΔQHCVertical(out-in)≈0, 而上游一般采用未受污染的背景地下水, 则QHCHorizontal(in)≈0, 计算公式(1)可简化为:
$$ R_{\text {Sat }}=Q_{\mathrm{HC}_{\text {Horizontal(out) }}}+Q_{\mathrm{H}_2 \mathrm{O}}\\ \left[ \begin{array}{l} \varSigma \left( {{S_{{\text{electron donor }}}}} \right.\Delta {C_{{\text{electron dono}}{{\rm{r}}_{{\rm{up - down}}}}{\rm{ }}}})\\ + \varSigma \left( {{S_{{\rm{byproduct }}}}} \right.\Delta {C_{{\rm{byproduc}}{{\rm{t}}_{{\rm{down - up}}}}{\rm{ }}}}) \end{array} \right] $$ (2) $$ \begin{array}{l} \varSigma \left( {{S_{{\text{electron donor }}}}} \right.\Delta {C_{{\text{electron dono}}{{\rm{r}}_{{\rm{up - down}}}}{\rm{ }}}}) = \\ {\left( {{S_{{{\rm{O}}_2}}}{C_{{{\rm{O}}_2}}} + {S_{{\rm{NO}}_3^ - }}{C_{{\rm{NO}}_3^ - }} + {S_{{\rm{SO}}_4^{2 - }}}C_{{\rm{SO}}_4^{2 - }}} \right)_{{\rm{up }}}} \end{array} $$ (3) $$ \begin{aligned} &\sum S_{\text {byproduct }} \Delta C_{\text {byproduct }}= \\ &\quad\left(S_{\mathrm{Fe}^{2+}} C_{\mathrm{Fe}^{2+}}+S_{\mathrm{Mn}^{2+}} C_{\mathrm{Mn}^{2+}}+S_{\mathrm{CH}_4} C_{\mathrm{CH}_4}\right)_{\text {down }} \end{aligned} $$ (4) 式中:QHCHorizontal(out)=-KAICHC。K为渗透系数(m/d); A为横截断面面积(m2); I为水力坡度, 无量纲; CHC为地下水中HC浓度(mg/L)。
4.3.2 包气带源区自然消除定量评估
目前, 包气带源区自然消除定量评估的常用方法分为三类: 浓度梯度法、二氧化碳通量法和热力学梯度法[26, 30, 49-54]。其中二氧化碳通量法也可分为动态密闭室法和静态捕集法。
以碳氢化合物为例, 包气带定量计算公式可表示为:
$$ R_{\text {Unsat }}=\int\int\left[J_{\mathrm{HC}}+S_{\mathrm{CH}_4} J_{\mathrm{CH}_4}+S_{\mathrm{O}_2} J_{\mathrm{O}_2}\right] \mathrm{d} x \mathrm{~d} y $$ (5) 式中:RUnsat为源区包气带中HC损失速率(kg-HC/d); JHC、SCH4JCH4、SO2JO2三项分别为包气带HC挥发、产甲烷、好氧降解导致的自然衰减通量(kg-HC/m2/d); dx、dy分别为源区的长、宽(m)。
(1) 浓度梯度法
浓度梯度法可视为源区自然消除定量评估的普适方法[15-24]。依据包气带源区自然消除概念模型和亨利定律, 通过在包气带不同深度布设气体监测井, 测定HC、CH4、O2等气体浓度, 定量评价包气带中HC损失速率:
$$ R_{\text {Unsat }}=\int\int\left[\begin{array}{l} -D_{\mathrm{HC}}\left(\frac{\partial C_{\mathrm{HC}}}{\partial z}\right)-S_{\mathrm{CH}_4} D_{\mathrm{CH}_4}\left(\frac{\partial C_{\mathrm{CH}_4}}{\partial z}\right) \\ +S_{\mathrm{O}_2} D_{\mathrm{O}_2}\left(\frac{\partial C_{\mathrm{O}_2}}{\partial z}\right) \end{array}\right] \mathrm{d} x \mathrm{~d} y $$ (6) 式中:DHC、DCH4、DO2分别为HC、CH4和O2的气体分子扩散系数(m2/d); $ \frac{\partial C_{\mathrm{HC}}}{\partial z}、\frac{\partial C_{\mathrm{CH}_4}}{\partial z}、\frac{\partial C_{\mathrm{O}_2}}{\partial z}$分别为由地下水面向上HC、CH4和O2的浓度变化(kg/m3/m)。
积分后, 公式(6)可表示为:
$$R_{\text {Unsat }}=A\left[\begin{array}{l} -D_{\mathrm{HC}}\left(\frac{\partial C_{\mathrm{HC}}}{\partial z}\right)-S_{\mathrm{CH}_4} D_{\mathrm{CH}_4}\left(\frac{\partial C_{\mathrm{CH}_4}}{\partial z}\right) \\ +S_{\mathrm{O}_2} D_{\mathrm{O}_2}\left(\frac{\partial C_{\mathrm{O}_2}}{\partial z}\right) \end{array}\right] $$ (7) 式中:A为源区面积(m2)。
研究发现, 一般情况下, 即使有HC和CH4经过甲烷氧化带挥发至地表, 其通量一般不到被氧气好氧降解生成二氧化碳的通量的1‰(可忽略不计)[15-24]。则计算公式(7)简化为:
$$R_{\text {Unsat }}=A S_{\mathrm{O}_2} D_{\mathrm{O}_2}\left(\frac{\partial C_{\mathrm{O}_2}}{\partial z}\right) $$ (8) 以Paul等在Guadalupe Oil Field污染场地的研究为例[18], 实地数据显示, 随着深度变化, O2明显减少, CO2明显增加, CH4和CH虽有检出但相对浓度较低。因此可以使用简化公式(8), 以O2为指标进行包气带源区自然消除速率的计算。以线性模型计算氧气浓度梯度变化, 即可求解得$\frac{\partial C_{\mathrm{O}_2}}{\partial z} $。扩散系数DO2取不同深度的均值, O2对HC的化学计量系数SO2为0.29kg-HC/kg-O2(表 4)。
据此以公式(8)计算, 可得到该污染场地各点位的包气带源区自然消除速率, 进而计算整个源区的包气带自然消除速率为2.1×105~6.3×105kg TPH /y, 而饱水带源区自然消除速率仅为1.1×103~3.9×103kg TPH/y, 比包气带源区自然消除速率低约两个数量级[18]。
(2) 二氧化碳通量法
与简化的浓度梯度法相比, 二氧化碳通量法应用的假设前提是: 达到地表的碳氢化合物(含甲烷)气体完全降解转化为二氧化碳(忽略HC和CH4)。包气带中HC损失速率为:
$$ R_{\text {Unsat }}=-A S_{\mathrm{CO}_2} D_{\mathrm{CO}_2}\left(\frac{\partial C_{\mathrm{CO}_2}}{\partial z}\right) $$ (9) 式中:SCO2为代谢产物CO2对HC的化学计量系数(kg-HC/kg-CO2); DCO2为CO2的气体分子扩散系数(m2/d)。
通过观测浅表土(一般不超过1m)的CO2, 即可通过地表CO2通量估算包气带中HC损失速率:
$$ R_{\text {Unsat }} \approx-A S_{\mathrm{CO}_2} J_{\mathrm{CO}_2} $$ (10) 式中:JCO2为地表土壤CO2通量(扣除土壤呼吸CO2通量,单位为kg/m2/d)。
根据观测的方式和装置不同, 二氧化碳通量法又分为静态捕集法和动态气室法两种(图 3)。其中, 静态捕集法的常用装置安装在地表, 可分别从顶部和底部以碱性物质吸收大气和土壤中的CO2, 获取土壤气CO2的通量, 计算HC损失速率。动态气室法直接将气室与CO2分析仪连接, 测量气室中CO2浓度的变化$ \frac{\partial C}{\partial t}$ (与大气温度T0、湿度W0、压强P0等相关), 获取土壤CO2通量并计算HC损失速率。二氧化碳通量法普遍用于碳循环土壤呼吸研究, 在此不再赘述。
![]() 图 3 二氧化碳通量法监测原理:(a) 静态捕集法; (b) 动态密闭室法Figure 3. Schematic illustration of CO2 monitoring method (a: CO2 trap method; b: dynamic closed chamber method)
图 3 二氧化碳通量法监测原理:(a) 静态捕集法; (b) 动态密闭室法Figure 3. Schematic illustration of CO2 monitoring method (a: CO2 trap method; b: dynamic closed chamber method)(3) 热力学梯度法
热力学梯度法是以地温估算源区自然消除的一种方法, 是一种较有前途的方法, 近些年相关研究明显增多[28, 30-31, 49]。但相较其他方法, 热力学梯度法在实际修复中的应用仍较少。该方法依据包气带源区自然消除概念模型, 认为包气带自然消除主要来源于产甲烷区和甲烷氧化区。产甲烷反应C10H22(aq)+4.5H2O(l)→2.25CO2(g)+7.75CH4(g)的吉布斯自由能ΔGr0为-266 kJ/mol-C10H22的放热反应, 甲烷氧化反应CH4(g)+2O2(g)→CO2(g)+2H2O(l)为吉布斯自由能ΔGr0为-6341kJ/mol-C10H22的放热反应, 两个反应联立C10H22(aq)+15.5O2(g)→10CO2(g)+11H2O(l)的总吉布斯自由能ΔGr0为-6607kJ/mol-C10H22[28, 30-31, 49]。通过合理观测不同位置和深度的地温变化, 可以包气带自然消除的效果进行估算。
5. 结论与展望
源区自然消除是对传统监控自然衰减概念的补充和深化。以往研究表明, 包气带源区自然消除效率大于饱水带源区自然消除, 包气带产甲烷和甲烷氧化是其发生的根源, 这些发现让人们对污染物自然衰减的时间周期有了新的认识, 对监控自然衰减修复的效率有了更高期望。虽然源区自然消除技术中的四分区概念模型已基本刻画清晰, 但目前的技术方法仅能实现通量监测, 尚未实现分区分过程的监测。对微生物的监测则并未涉及, 机理过程的揭示尚有不足。
在污染场地修复过程中, 类似于地下水中添加电子受体强化自然衰减一样, 要将源区自然消除发展成为一种适用于实际需求的修复技术并能推广应用, 还需要对一系列科学问题开展深入研究, 主要包括: ①查明源区自然消除过程中LNAPL发生降解的主要组分, 以使概念模型更为具体化。②建立观测评估甲烷和二氧化碳的直接脱气和气泡逃逸方法, 以尽量避免结果的不确定性。③揭示源区自然消除的速率限速因子, 为强化措施的实施提供科学依据。
另外, 在观测评估方法方面, 虽然文献中在场地尺度对三种方法的定量评估结果作了详细对比[30, 50], 但对于哪种方法能更真实地表征源区自然消除尚无明确结论。进一步研究中, 需要基于概念模型开展大型物理仿真模拟试验, 在合适的尺度上模拟饱水带-包气带源区自然消除的四个分区, 以建立分区评估技术方法。
综上, 源区自然消除是污染修复领域一个新的研究方向。当前的研究成果为非水相液体污染场地修复和管理提供了新视角, 但与此相关的一系列基础科学与技术问题, 还需要更深入的研究。
-
![]()
![]()
图 2 息烽地区陡山沱组磷块岩的岩石学特征
a、b —球粒状磷块岩,分别为单偏光和反射光;c—似胚胎生物化石磷块岩,正交偏光;d—似胚胎生物化石,外膜被局部破坏,单偏光;e、f—似胚胎生物化石,球粒内部出现内陷和褶皱现象,有分裂生长,分别为单偏光和正交偏光。
Figure 2. Petrological characteristics of phosphorite from Doushantuo Formation in Xifeng area
![]()
![]()
表 1 息烽地区陡山沱组PAS硫同位素组成数据
Table 1 Sulfur isotopic composition data of PAS of Doushantuo Formation in Xifeng area
样品编号 PAS硫同位素数据δ34S(‰) 样品编号 PAS硫同位素数据δ34S(‰) XF-1 33.8 XF-17 32.8 XF-2 33.0 XF-18 33.9 XF-3 33.5 XF-19 32.9 XF-4 33.9 XF-20 33.2 XF-5 33.7 XF-21 33.8 XF-6 36.9 XF-22 34.3 XF-7 35.7 XF-23 34.2 XF-8 34.2 XF-24 35.3 XF-9 34.3 XF-25 35.1 XF-10 35.2 XF-26 34.4 XF-11 33.0 XF-27 34.1 XF-12 32.7 XF-28 34.2 XF-13 34.7 XF-29 34.3 XF-14 33.8 XF-30 34.3 XF-15 35.4 XF-31 32.7 XF-16 34.4 XF-32 34.4  下载: 导出CSV
下载: 导出CSV
-
[1] 张亚冠, 杜远生, 刘建中, 等. 贵州震旦系陡山沱组磷块岩成磷作用及与新元古代末期氧化事件(NOE)的耦合[J]. 古地理学报, 2020, 22(5): 893-912. https://www.cnki.com.cn/Article/CJFDTOTAL-GDLX202005007.htm Zhang Y G, Du Y S, Liu J Z, et al. Phosphogenesis of phosphorite from the Sinian Doushantuo Formation in Guizhou Province and its coupling relation with the Neoproterozoic Oxygenation Event (NOE)[J]. Journal of Palaeogeography (Chinese Edition), 2020, 22(5): 893-912. https://www.cnki.com.cn/Article/CJFDTOTAL-GDLX202005007.htm
[2] Filippelli G M. Phosphate rock formation and marine phosphorus geochemistry: The deep time perspective[J]. Chemosphere, 2011, 84(6): 759-766. doi: 10.1016/j.chemosphere.2011.02.019
[3] Anderson R P, Macdonald F A, Jones D S, et al. Doushantuo-type microfossils from latest Ediacaran phosphorites of northern Mongolia[J]. Geology, 2017, 45(12): 1079-1082. doi: 10.1130/G39576.1
[4] Pufahl P K, Hiatt E E. Oxygenation of the Earth's atmosphere-ocean system; a review of physical and chemical sedimentologic responses[J]. Marine and Petroleum Geology, 2012, 32(1): 1-20. doi: 10.1016/j.marpetgeo.2011.12.002
[5] Pufahl P K, Groat L A. Sedimentary and igneous phosphate deposits; formation and exploration; an invited paper[J]. Economic Geology and the Bulletin of the Society of Economic Geologists, 2017, 112(3): 483-516. doi: 10.2113/econgeo.112.3.483
[6] Hiatt E E, Pufahl P K, Edwards C T. Sedimentary phosphate and associated fossil bacteria in a Paleoproterozoic tidal flat in the 1.85Ga Michigamme Formation, Michigan, USA[J]. Sedimentary Geology, 2015, 319: 24-39. doi: 10.1016/j.sedgeo.2015.01.006
[7] Caird R A, Pufahl P K, Hiatt E E, et al. Ediacaran stromatolites and intertidal phosphorite of the Salitre Formation, Brazil; phosphogenesis during the Neoproterozoic Oxygenation Event[J]. Sedimentary Geology, 2017, 350: 55-71. doi: 10.1016/j.sedgeo.2017.01.005
[8] 王志罡, 谢宏, 杨旭, 等. 贵州铜仁坝黄磷矿中铀赋存状态的逐级化学提取研究[J]. 岩矿测试, 2018, 37(3): 256-265. doi: 10.15898/j.cnki.11-2131/td.201710310172 Wang Z G, Xie H, Yang X, et al. Stepwise extraction study on the occurrence of uranium in Tongren Bahuang Phosphorite, Guizhou[J]. Rock and Mineral Analysis, 2018, 37(3): 256-265. doi: 10.15898/j.cnki.11-2131/td.201710310172
[9] Baturin G N. The origin of marine phosphorites[J]. International Geology Review, 1989, 31(4): 327-342. doi: 10.1080/00206818909465885
[10] 张亚冠, 杜远生, 陈国勇, 等. 富磷矿三阶段动态成矿模式: 黔中开阳式高品位磷矿成矿机制[J]. 古地理学报, 2019, 21(2): 351-368. https://www.cnki.com.cn/Article/CJFDTOTAL-GDLX201902011.htm Zhang Y G, Du Y S, Chen G Y, et al. Three stages dynamic mineralization model of the phosphate-rich deposits: Mineralization mechanism of the Kaiyang-type high-grade phosphorite in central Guizhou Province[J]. Journal of Palaeogeography (Chinese Edition), 2019, 21(2): 351-368. https://www.cnki.com.cn/Article/CJFDTOTAL-GDLX201902011.htm
[11] Goldberg T, Poulton S W, Strauss H. Sulphur and oxygen isotope signatures of late Neoproterozoic to early Cambrian sulphate, Yangtze Platform, China: Diagenetic constraints and seawater evolution[J]. Precambrian Research, 2005, 137(3-4): 223-241. doi: 10.1016/j.precamres.2005.03.003
[12] Qiao W L, Lang X G, Peng Y B, et al. Sulfur and oxygen isotopes of sulfate extracted from early Cambrian phosphorite nodules: Implications for marine redox evolution in the Yangtze Platform[J]. Journal of Earth Science (Wuhan, China), 2016, 27(2): 170-179.
[13] Zhu M Y, Zhang J M, Yang A H. Integrated Ediacaran (Sinian) chronostratigraphy of South China[J]. Palaeogeography, Palaeoclimatology, Palaeoecology, 2007, 254(1-2): 7-61. doi: 10.1016/j.palaeo.2007.03.025
[14] 刘静江, 李伟, 张宝民, 等. 上扬子地区震旦纪沉积古地理[J]. 古地理学报, 2015, 17(6): 735-753. https://www.cnki.com.cn/Article/CJFDTOTAL-GDLX201506002.htm Liu J J, Li W, Zhang B M, et al. Sedimentary palaeo-geography of the Sinian in Upper Yangtze Region[J]. Journal of Palaeogeography (Chinese Edition), 2015, 17(6): 735-753. https://www.cnki.com.cn/Article/CJFDTOTAL-GDLX201506002.htm
[15] Jiang G Q, Shi X Y, Zhang S H, et al. Stratigraphy and paleogeography of the Ediacaran Doushantuo Formation (ca. 635-551Ma) in South China[J]. Gondwana Research, 2011, 19(4): 831-849. doi: 10.1016/j.gr.2011.01.006
[16] 杨爱华, 朱茂炎, 张俊明, 等. 扬子板块埃迪卡拉系(震旦系)陡山沱组层序地层划分与对比[J]. 古地理学报, 2015, 17(1): 1-20. https://www.cnki.com.cn/Article/CJFDTOTAL-GDLX201501001.htm Yang A H, Zhu M Y, Zhang J M, et al. Sequence stratigraphic subdivision and correlation of the Ediacaran (Sinian) Doushantuo Formation of Yangtze Plate, South China[J]. Journal of Palaeogeography (Chinese Edition), 2015, 17(1): 1-20. https://www.cnki.com.cn/Article/CJFDTOTAL-GDLX201501001.htm
[17] 徐丽, 邢蓝田, 王鑫, 等. 元素分析仪-同位素比值质谱测量碳氮同位素比值最佳反应温度和进样量的确定[J]. 岩矿测试, 2018, 37(1): 15-20. doi: 10.15898/j.cnki.11-2131/td.201701130005 Xu L, Xing L T, Wang X, et al. Study on the optimal reaction temperature and sampling weight for measurement of carbon and nitrogen isotope ratio by elemental analyzer-isotope ratio mass spectrometer[J]. Rock and Mineral Analysis, 2018, 37(1): 15-20. doi: 10.15898/j.cnki.11-2131/td.201701130005
[18] 高建飞, 徐衍明, 范昌福, 等. 元素分析仪-气体同位素质谱法分析硫酸钙样品的硫同位素组成[J]. 岩矿测试, 2020, 39(1): 53-58. doi: 10.15898/j.cnki.11-2131/td.201908120128 Gao J F, Xu Y M, Fan C F, et al. Analysis of sulfur isotope composition of gypsum samples by elemental analyzer-isotope mass spectrometry[J]. Rock and Mineral Analysis, 2020, 39(1): 53-58. doi: 10.15898/j.cnki.11-2131/td.201908120128
[19] Paytan A, Kastner M, Campbell D, et al. Sulfur isotopic composition of Cenozoic seawater sulfate[J]. Science, 1998, 282(5393): 1459-1462. doi: 10.1126/science.282.5393.1459
[20] Gill B C, Lyons T W, Jenkyns H C. A global perturbation to the sulfur cycle during the Toarcian Oceanic Anoxic Event[J]. Earth and Planetary Science Letters, 2011, 312(3-4): 484-496. doi: 10.1016/j.epsl.2011.10.030
[21] Crockford P W, Kunzmann M, Bekker A, et al. Claypool continued: Extending the isotopic record of sedimentary sulfate[J]. Chemical Geology, 2019, 513: 200-225. doi: 10.1016/j.chemgeo.2019.02.030
[22] Fike D A, Bradley A S, Rose C V. Rethinking the ancient sulfur cycle[J]. Annual Review of Earth and Planetary Sciences, 2015, 43: 593-622. doi: 10.1146/annurev-earth-060313-054802
[23] Gomes M L, Hurtgen M T. Sulfur isotope fractionation in modern euxinic systems: Implications for paleo-environmental reconstructions of paired sulfate-sulfide isotope records[J]. Geochimica et Cosmochimica Acta, 2015, 157: 39-55. doi: 10.1016/j.gca.2015.02.031
[24] Wang W, Guan C, Zhou C, et al. Integrated carbon, sulfur, and nitrogen isotope chemostratigraphy of the Ediacaran Lantian Formation in South China: Spatial gradient, ocean redox oscillation, and fossil distribution[J]. Geobiology, 2017, 15(4): 552-571. doi: 10.1111/gbi.12226
[25] Bristow T F, Grotzinger J P. Sulfate availability and the geological record of cold-seep deposits[J]. Geology, 2013, 41(7): 811-814. doi: 10.1130/G34265.1
[26] Present T M, Gutierrez M, Paris G, et al. Diagenetic controls on the isotopic composition of carbonate-associated sulphate in the Permian Capitan Reef Complex, West Texas[J]. Sedimentology, 2019, 66(7): 2605-2626. doi: 10.1111/sed.12615
[27] Horacek M, Brandner R, Richoz S, et al. Lower Triassic sulphur isotope curve of marine sulphates from the Dolomites, N-Italy[J]. Palaeogeography, Palaeocli-matology, Palaeoecology, 2010, 290(1-4): 65-70. doi: 10.1016/j.palaeo.2010.02.016
[28] Prince J, Rainbird R H, Wing B A. Evaporite deposition in the mid-Neoproterozoic as a driver for changes in seawater chemistry and the biogeochemical cycle of sulfur[J]. Geology, 2019, 47(4): 375-379. doi: 10.1130/G45464.1
[29] Horita J, Zimmermann H, Holland H D. Chemical evolution of seawater during the Phanerozoic: Implications from the record of marine evaporites[J]. Geochimica et Cosmochimica Acta, 2002, 66(21): 3733-3756. doi: 10.1016/S0016-7037(01)00884-5
[30] Paytan A, Mearon S, Cobb K M, et al. Origin of marine barite deposits: Sr and S isotope characterization[J]. Geology, 2002, 30(8): 747-750. doi: 10.1130/0091-7613(2002)030<0747:OOMBDS>2.0.CO;2
[31] Griffith E M, Paytan A. Barite in the ocean-occurrence, geochemistry and palaeoceanographic applications[J]. Sedimentology, 2012, 59(6): 1817-1835. doi: 10.1111/j.1365-3091.2012.01327.x
[32] Paris G, Adkins J F, Sessions A L, et al. Neoarchean carbonate-associated sulfate records positive Δ33S anomalies[J]. Science, 2014, 346(6240): 739-741.
[33] Tostevin R, He T C, Turchyn A V, et al. Constraints on the late Ediacaran sulfur cycle from carbonate associated sulfate[J]. Precambrian Research, 2017, 290: 113-125. doi: 10.1016/j.precamres.2017.01.004
[34] Paris G, Fehrenbacher J S, Sessions A L, et al. Experimental determination of carbonate-associated sulfate δ34S in planktonic foraminifera shells[J]. Geochemistry, Geophysics, Geosystems, 2014, 15(4): 1452-1561. doi: 10.1002/2014GC005295
[35] Ma H R, Dong L, Shen B, et al. Sulfur and oxygen isotopic compositions of carbonate associated sulfate (CAS) of Cambrian ribbon rocks: Implications for the constraints on using CAS to reconstruct seawater sulfate sulfur isotopic compositions[J]. Chemical Geology, 2021, 580: 120369. doi: 10.1016/j.chemgeo.2021.120369
[36] Wu N, Farquhar J, Fike D A. Ediacaran sulfur cycle: Insights from sulfur isotope measurements (Δ33S and δ34S) on paired sulfate-pyrite in the Huqf Supergroup of Oman[J]. Geochimica et Cosmochimica Acta, 2015, 164: 352-364. doi: 10.1016/j.gca.2015.05.031
[37] Wang R M, Lang X G, Ding W M, et al. The coupling of Phanerozoic continental weathering and marine phosphorus cycle[J]. Scientific Reports, 2020, 10(1): 5794. doi: 10.1038/s41598-020-62816-z
[38] Hawkings J, Wadham J, Tranter M, et al. The Greenland Ice Sheet as a hot spot of phosphorus weathering and export in the Arctic[J]. Global Biogeochemical Cycles, 2016, 30(2): 191-210. doi: 10.1002/2015GB005237
[39] Compton J, Mallinson D, Glenn C R, et al. Variations in the global phosphorus cycle[J]. Special Publication-Society for Sedimentary Geology, 2000, 66: 21-33.
[40] Zhenbing S, Strother P, Papineau D. Terminal Proterozoic cyanobacterial blooms and phosphogenesis documented by the Doushantuo granular phosphorites Ⅱ: Microbial diversity and C isotopes[J]. Precambrian Research, 2014, 251: 62-79. doi: 10.1016/j.precamres.2014.06.004
[41] Cui H, Xiao S, Chuanming Z, et al. Phosphogenesis asso-ciated with the Shuram Excursion; petrographic and geochemical observations from the Ediacaran Doushantuo Formation of South China[J]. Sedimentary Geology, 2016, 341: 134-146. doi: 10.1016/j.sedgeo.2016.05.008
[42] Nelson G J, Pufahl P K, Hiatt E E. Paleoceanographic constraints on Precambrian phosphorite accumulation, Baraga Group, Michigan, USA[J]. Sedimentary Geology, 2010, 226(1-4): 9-21. doi: 10.1016/j.sedgeo.2010.02.001
[43] Canfield D E, Poulton S W, Knoll A H, et al. Ferruginous conditions dominated later Neoproterozoic deep-water chemistry[J]. Science, 2008, 321(5891): 949-952. doi: 10.1126/science.1154499
[44] Li C, Love G D, Lyons T W, et al. A stratified redox model for the Ediacaran Ocean[J]. Science, 2010, 328(5974): 80-83. doi: 10.1126/science.1182369
[45] Johnston D T, Poulton S W, Dehler C, et al. An emerging picture of Neoproterozoic ocean chemistry: Insights from the Chuar Group, Grand Canyon, USA[J]. Earth and Planetary Science Letters, 2010, 290(1-2): 64-73. doi: 10.1016/j.epsl.2009.11.059
[46] Li C, Cheng M, Algeo T J, et al. A theoretical prediction of chemical zonation in early oceans (>520Ma)[J]. Science China: Earth Sciences, 2015, 58(11): 1901-1909. doi: 10.1007/s11430-015-5190-7
[47] Raiswell R, Canfield D E. The iron biogeochemical cycle past and present[J]. Geochemical Perspectives, 2012, 1(1): 1-220. doi: 10.7185/geochempersp.1.1
-
期刊类型引用(4)
1. 李飞,郭家泽,杨永平,朱攀,陈金伟,段文. 昆明市碧鸡街道某村土壤样品中碘含量的测定. 化工设计通讯. 2025(01): 121-123 .  百度学术
百度学术
2. 黄锐敏,吴智威,郑云云,黄崇耀. 氟化氢铵消解-甲烷增敏-电感耦合等离子体质谱法测定土壤中的碘. 分析科学学报. 2024(01): 105-110 . 百度学术
3. 鲁银鹏,孟郁苗,黄小文,王丛林,杨秉阳,谭侯铭睿,谢欢. 宁芜盆地玢岩型铁矿尾矿元素与矿物组成特征. 岩矿测试. 2024(02): 259-269 . 本站查看
4. 李月娥,孟庆俊,李昌平,刘淼,徐辉,谢楠,曹恩伟,张晓赟,蒋思丝,顾晓明,曹珣. 电感耦合等离子体质谱(ICP-MS)法测定土壤中的碘. 中国无机分析化学. 2023(06): 549-555 . 百度学术
其他类型引用(0)




计量
- 文章访问数: 287
- HTML全文浏览量: 68
- PDF下载量: 33
- 被引次数: 4
 京公网安备 11010202008159号
京公网安备 11010202008159号