
Determination of 8 Organic Pollutants in Drinking Waters by Ultra-high Performance Liquid Chromatography Tandem Mass Spectrometry with Direct Injection
-
摘要:
2011年我国实施了新的生活饮用水卫生标准,卫生指标扩展至106项,并配套了相关的检测方法,其中微囊藻毒素、莠去津采用液相色谱紫外检测器的检测方法;丙烯酰胺、灭草松、2,4-D采用气相色谱-电子捕获检测器的检测方法;呋喃丹、草甘膦采用液相色谱荧光检测器的检测方法。这些检测方法均有前处理程序繁琐、工作量大、可操作性差的缺点,在实际工作中应用较为困难。本文采用超高效液相色谱-串联质谱技术(UPLC-MS/MS),建立了水样过0.22 μm滤膜,直接进样测定自来水、地下水及地表水中8种有机污染物(草甘膦、呋喃丹、灭草松、莠去津、2, 4-滴、丙烯酰胺、微囊藻毒素-RR和微囊藻毒素-LR)的分析方法。8种化合物在各自的线性范围内线性关系良好,相关系数均不低于0.993,定量下限为0.07~5 μg/L,低、中、高三个加标水平的回收率为94.2%~103.7%,相对标准偏差为1.1%~7.8%。本文建立的方法操作简便,可以实现在6 min内一次性分析8种目标化合物,与国家标准方法相比,显著提高了分析效率;与文献报道的均是单一类化合物检测方法相比,本检测方法具有相似的灵敏度和检测限,但是目标化合物覆盖更全面,分析时间更短。
-
关键词:
- 水样 /
- 农药 /
- 丙烯酰胺 /
- 微囊藻毒素 /
- 超高效液相色谱-串联质谱法
Abstract:In 2011, new healthy standards for drinking water were implemented, which included 106 health indicators and related detection methods. In the national standard methods, microcystins and atrazine are detected by Liquid Chromatography with Ultraviolet Detector; acrylamide, bentazone and 2,4-D are detected by Gas Chromatography with Electron Capture Detector, carbofuran and glyphosate are measured by Liquid Chromatography with Fluorescence Detector. All of these methods are cumbersome and have poor operability making them difficult to conduct on a daily basis. A method for the analysis of 8 organic pollutants (glyphosate, carbofuran, bentazone, atrazine, 2,4-D, acrylamide, microcystin-RR and microcystin-LR) in drinking water, surface water and underground water was developed using Ultra Performance Liquid Chromatography with Tandem Mass Spectrometry (UPLC-MS-MS). After filtration by 0.22 μm membrane, the water sample was injection directly into the equipment. Compared with recently reported methods, similar precision and accuracy were obtained and are presented in this paper. But shorter analysis time is needed and more target compounds are included. The result indicate that the LOQ of 8 compounds ranged from 0.07 to 5 μg/L, Meanwhile, the high correlation coefficients (r>0.993) of 8 compounds were obtained within the respective linear ranges. The average recoveries at low, intermediate and high spiked levels range from 96.7% to 101.7% with relative standard deviations (RSD, n=6) of 1.1%-3.5%. This method is suitable for the identification and quantification of the aforementioned 8 compounds in drinking water with the advantages of simple pretreatment, good reliability and high sensitivity.
-
在宝石学中,主要由低温石英(α-石英)微晶组成且具有一定工艺价值的矿物集合体通常被称为石英质玉石。该类玉石分布十分广泛,并凭借其多彩的颜色、温润细腻的质地而深受人们青睐,具有较高的市场潜力,从而推动了石英质玉石的相关研究。石英质玉石的成分往往对其外观特征、品质级别、应用性能等方面有着显著影响,因此该领域也是石英质玉石研究中的热点。随着研究的不断深入,人们发现石英质玉石中还可存在一种低温低压SiO2矿物——斜硅石[1]。1976年斜硅石首次发现于西班牙大加纳利岛(Gran Canaria)Mogan组底层流纹质凝灰岩的裂隙和孔洞中[2],其晶体结构可以看作通过左形α-石英与右形α-石英的(1011) 面网交替堆叠,形成一个由SiO4四面体共角连接而成的三维结构,在单个晶胞范围内这种结构类似于作周期性重复的巴西双晶[3]。2007年国际矿物学会新矿物和矿物命名委员会正式批准,将斜硅石作为一种具有与石英相似的AB2结构的新矿物种(CNMMN No.99-035) 列入矿物名录。斜硅石和α-石英可共存于常温常压环境中,但两者具有不同的晶体结构:斜硅石的晶胞参数a=0.8758 nm,b=0.4876 nm,c=1.0715 nm,β=90.08°,其空间群为I2/a,属单斜晶系(由于β=90.08°,外观常呈假斜方对称),其对称性低于α-石英(α-石英的晶胞参数a=0.4913 nm,b=0.4913 nm,c=0.5404 nm,空间群为P3121,属三方晶系)[2, 3]。
斜硅石常以纳米尺度微晶的形式存在于各种产状、各种颜色以及不同结晶程度的石英质玉石中[1, 3, 4, 5],仅凭肉眼观察一般无法将其鉴别出来。迄今为止,斜硅石的研究主要涉及晶体结构[2, 3, 6, 7]、形态特征[1, 4, 5]、物理化学性质[1, 3]、检测方法[8, 9, 10, 11, 12, 13, 14, 15]、分布特征[1, 7, 16]和形成机理[17, 18, 19, 20, 21, 22]等方面,但关于斜硅石的相对含量对石英质玉石结晶程度的影响还未见报道,开展这些方面的研究将为大致判断石英质玉石的结晶程度提供新思路,也可为石英质玉石的品质评价、综合利用等建立科学依据和理论约束。
本文选取中国云南龙陵、安徽大别山地区、广西贺州、内蒙古阿拉善盟地区4个代表性产地所产出的不同颜色、不同结构类型的石英质玉石为研究对象,分别利用拉曼光谱、红外光谱、X射线粉晶衍射三种宝玉石研究中较为常用的测试方法,探讨了石英质玉石中斜硅石和α-石英的相对含量与石英质玉石结晶程度之间的关系,并结合斜硅石与α-石英的晶体结构特征给予了理论上的解释。
1. 实验部分
1.1 实验样品
本次研究的19件样品是从100件来自不同产地(云南龙陵、安徽大别山地区、广西贺州和内蒙古阿拉善盟地区)的石英质玉石中挑选出来的典型样品,通过偏光显微镜对样品磨制的岩石薄片进行分析,研究样品的显微结构可分为粒状结构和纤维状结构两大类。
1.2 样品分析方法及其测量条件
(1) 拉曼光谱分析:采用Renishaw-inVia型激光共聚焦显微拉曼光谱仪(英国雷尼绍公司)。仪器测量条件为:激发光源波长532 nm,分辨率1 cm-1,激光功率10 mW,测量范围100~1200 cm-1,扫描次数为3次,对测试图谱进行峰形拟合所使用的软件为Wire 4.1。
(2) 红外漫反射光谱分析:采用NICOLET iS5傅里叶变换红外光谱仪(美国尼高力公司)的PIKE TECHNOLOGIES UplRTM红外漫反射附件对19件样品进行测试,所有测试图谱均进行K-K变换予以校正。仪器主要测量参数为:扫描范围400~2000 cm-1,扫描次数为32次,分辨率4 cm-1。
(3) X射线衍射分析:采用Bruker D8 Advance型6.5kW转靶X射线衍射仪(美国Bruker公司)进行测试。根据研究内容的不同,测量参数设置略有差异:在物相鉴定时测量角度范围为10°~80°,扫描速度4°/min,步长0.02°;而在结晶度测量时角度范围为67°~69°,扫描速度0.25°/min。为尽可能地减少晶体粒度和择优取向对测量结果的影响,将样品均磨制成粒度为74 μm(200目)以下的粉末。
2. 结果与讨论
2.1 斜硅石及其相对含量表征
2.1.1 拉曼光谱分析
前人研究表明,斜硅石具有19个独立的拉曼光谱谱峰,其中多数与α-石英重合,最特征的也是最强的斜硅石拉曼特征峰位于502 cm-1附近,该峰主要与硅氧四面体组成的四方环中Si—O—Si的对称伸缩-弯曲振动有关,因此常用于确定是否有斜硅石存在[9, 12, 23]。由于石英质玉石中常含有黄色、红色等主要由铁的氧化物(氢氧化物)组成的致色物质,这些杂质矿物会对斜硅石的拉曼光谱产生影响,如赤铁矿在496 cm-1附近具有特征峰。因此,本次研究选取无色或接近无色的样品进行拉曼光谱测试,结果显示不同样品所包含的502 cm-1谱峰的强度差异较大,说明不同样品中斜硅石的相对含量存在差异。样品中的斜硅石含量越高,其拉曼光谱中斜硅石502 cm-1特征峰就越为明显。
斜硅石含量的多少可以根据研究样品的拉曼光谱和X射线粉晶衍射图谱的分析和计算获得。GÖtze等(1998)[9]配制了不同含量比例的斜硅石/α-石英样品,对其进行拉曼光谱测试,发现斜硅石在样品中的相对含量越高,拉曼光谱中502 cm-1/465 cm-1谱峰面积的比值和峰强度的比值也越高。本文利用高斯函数及洛伦兹函数对502 cm-1和465 cm-1进行峰形拟合,分别求出两峰的积分面积比(A502/A465,%)和强度比(I502/I465,%), 见表 1。有必要说明的是,由于部分样品的502 cm-1特征峰较小,无法进行较好的峰形拟合,从而计算得出的积分面积比的误差也相对较大,因此表 1中仅列出了这些样品的两峰强度比I502/I465。
表 1 不同产地石英质玉石样品的拉曼光谱和红外漫反射光谱信息Table 1. Data of Raman spectra and infrared diffuse reflectance spectra of quartzite jade samples from different geographic origins样品产地 样品编号 结构
类型拉曼光谱
峰强度比
I502/I465(%)拉曼光谱
峰面积比
A502/A465(%)红外光谱
强度比
I1095/I1157内蒙古
阿拉善盟
地区ALS-1 纤维状 15.29 15.87 1.746 ALS-2 纤维状 17.27 18.54 1.789 ALS-CH-1 纤维状 16.25 16.47 1.749 ALS-MNS-1 纤维状 19.54 19.63 1.874 ALS-MNS-2 纤维状 14.68 15.24 1.772 ALS-MNS-3 纤维状 16.00 16.13 1.743 ALS-BLMN-1 纤维状 16.54 17.21 1.759 ALS-BLMN-2 纤维状 20.37 22.00 1.724 ALS-KD1-1 纤维状 16.19 16.35 1.837 ALS-KD1-2 粒状 15.30 15.94 1.783 ALS-MNT-1 粒状 12.75 12.50 1.714 ALS-MNT-2 粒状 0 0 1.508 安徽霍山 HSY-60 粒状 2.92 - 1.722 TB1235 粒状 0 0 1.628 广西贺州 HZ-C-030 粒状 2.54 - 1.512 市场购买
(产地不详)AGATE-20 纤维状 16.30 17.62 1.793 AGATE-21 纤维状 17.13 17.93 1.830 AGAM10018 纤维状 4.62 - 1.602 AGAM10019 纤维状 0 0 1.605 注:I502/I465和A502/A465的数据均为每件样品20个测试点的平均
值。个别样品由于502 cm-1特征峰过小,无法准确计算出峰面
积,在表中以“-”表示。将表 1中可进行峰形拟合的样品的I502/I465和A502/A465进行对比可知,两种计算方法得出的结果均相对偏差在3%以内,并且其变化趋势基本相同。因此,考虑到研究样品中的斜硅石含量较低,对斜硅石的502 cm-1特征峰较小的部分样品进行峰形拟合时存在一定困难且误差较大,本文将以拉曼光谱的502 cm-1/465 cm-1峰强度比I502/I465来表示斜硅石含量相对大小的变化。
2.1.2 红外漫反射光谱分析
斜硅石在石英质玉石中的相对含量变化同样可对红外漫反射光谱产生影响。Hardgrove(2013)[12]对含有不同比例斜硅石的石英质玉石样品进行红外漫反射光谱分析,发现1095 cm-1与1157 cm-1两处谱线强度比(记作I1095/I1157)会随着斜硅石含量的改变而发生明显的变化。本文对19件研究样品进行红外漫反射光谱测试并计算其红外漫反射光谱强度比值 I1095/I1157(表 1),再将样品测试数据投点绘制成“红外漫反射光谱 I1095/I1157与拉曼光谱I502/I465散点分布图”(图 1)。由图 1可知,当拉曼光谱中的502 cm-1与465 cm-1峰强度比I502/I465>12%时,红外漫反射光谱中的I1095/I1157比值与其呈较明显的正相关关系;而当502 cm-1与465 cm-1两峰的强度比I502/I465<5%时,两者的相关性较弱。
![]() 图 1 红外漫反射光谱中I1095/I1157与拉曼光谱中I502/I465之间的关系Figure 1. The relationship between I1095/I1157 of infrared diffuse reflection spectrum and I502/I465 of Raman spectrum
图 1 红外漫反射光谱中I1095/I1157与拉曼光谱中I502/I465之间的关系Figure 1. The relationship between I1095/I1157 of infrared diffuse reflection spectrum and I502/I465 of Raman spectrum2.1.3 X射线衍射分析
斜硅石的主要特征d值为4.43、4.35、3.11、2.88[7]。根据布拉格公式λ=2dsinθ,上述各d值对应的2θ分别为19.92°、20.27°、28.72°、30.97°。
根据拉曼光谱和红外漫反射光谱的测试结果,选取7件斜硅石相对含量不同的典型样品切除围岩并研磨成74 μm以下的粉末,进行X射线粉晶衍射测试(图 2)。由图 2可知,样品中斜硅石的衍射峰强度普遍较弱,说明样品中斜硅石的含量普遍较低。一些研究[9, 12]通过对比两种计算结果发现,拉曼光谱测试所获得的斜硅石含量更接近于真实值,并认为这种差异可能是因为X射线衍射对于纳米尺度的斜硅石无法产生连续的布拉格散射,而拉曼光谱的微小光斑对纳米尺度的斜硅石的短程有序结构反应灵敏所致。由于样品的X射线衍射分析获得的斜硅石的衍射峰普遍较弱,因此本文主要采用拉曼光谱的计算结果来表征样品中斜硅石相对含量的变化。
![]() 图 2 不同斜硅石含量样品的X射线粉晶衍射图谱:(a)斜硅石衍射峰未出现或不明显; (b)斜硅石衍射峰较为明显Q—α-石英衍射峰; M—斜硅石衍射峰。Figure 2. XRD patterns of typical samples with different moganite contents: (a) XRD patterns with less obvious or without diffraction peaks of moganite; (b) XRD patterns with relatively obvious diffraction peaks of moganite
图 2 不同斜硅石含量样品的X射线粉晶衍射图谱:(a)斜硅石衍射峰未出现或不明显; (b)斜硅石衍射峰较为明显Q—α-石英衍射峰; M—斜硅石衍射峰。Figure 2. XRD patterns of typical samples with different moganite contents: (a) XRD patterns with less obvious or without diffraction peaks of moganite; (b) XRD patterns with relatively obvious diffraction peaks of moganite2.2 斜硅石相对含量与石英质玉石结晶度的关系
X射线衍射技术同样可以用来测量矿物的结晶度。结晶度可以描述为结晶的完全程度或完整程度,其中结晶的完全程度是指物质中非晶体与晶体的相对比例,而结晶的完整程度是指物质中内部质点排列的规则程度以及结构缺陷的多少[24]。本次研究样品的X射线粉晶衍射图谱中无非晶体散射峰存在,所以本文所指的结晶度主要是石英质玉石结晶完整程度的体现。已有研究[25, 26]表明,X射线粉晶衍射图谱中67°~69°范围内的五指衍射峰可用于计算石英质玉石的结晶度指数(CI),计算公式为:CI=10·F·a/b。式中的a、b值可由位于67.74°附近、对应d(2132)=0.13844 nm衍射峰测量得出(图 3);F为比例因子,不同衍射仪的F值一般不同,需要用标准样品进行标定(一般为纯净无杂质的人工水晶,其CI值为10)。本文所选用的标准样品是由一个无色透明、内部基本无任何包裹体的水晶研磨而成的粉末。根据上述公式计算,得出本次结晶度计算公式中比例因子F=1.179。7件典型样品的结晶度指数见图 4中各点的纵坐标。
![]() 图 3 7件测试样品XRD图谱的68°五指衍射峰Figure 3. The 68° quintuplet peaks in the XRD patterns of seven investigated samples
图 3 7件测试样品XRD图谱的68°五指衍射峰Figure 3. The 68° quintuplet peaks in the XRD patterns of seven investigated samples结合拉曼光谱和X射线粉晶衍射的计算结果,分别对7件X射线衍射测试样品的502 cm-1/465 cm-1谱峰强度比I502/I465和石英质玉石结晶度指数进行投点,绘制了“I502/I465-石英结晶度指数”散点分布图(图 4)。结果显示,无论是粒状结构还是纤维状结构的石英质玉石,其中斜硅石的相对含量与样品结晶度的关系有着共同的规律,即502 cm-1/465 cm-1峰强度比值(I502/I465)越大,斜硅石的相对含量越高,样品的结晶度越低。通过Excel软件对数据进行线性回归分析,石英质玉石的结晶度指数(Y)与502 cm-1/465 cm-1峰强度比值(X)之间的变化规律大致符合负相关关系:Y=-0.36X+6.93,相关系数r=-0.94。
![]() 图 4 拉曼光谱中峰强度比值I502/I465(%)与石英质玉石结晶度(CI)的关系Figure 4. The relationship between the peak intensity ratio I502/I465(%) of Raman spectrum and the crystallinity index of quartzite jade
图 4 拉曼光谱中峰强度比值I502/I465(%)与石英质玉石结晶度(CI)的关系Figure 4. The relationship between the peak intensity ratio I502/I465(%) of Raman spectrum and the crystallinity index of quartzite jade2.3 斜硅石相对含量对石英质玉石结晶度影响的理论解释
石英质玉石中斜硅石的相对含量与石英质玉石结晶度之间的负相关关系,可以从晶体结构的角度进行解释。斜硅石的晶体结构可以看作是由左形α-石英与右形α-石英的(1011) 面晶格面网平行斜硅石晶体(101) 面交替堆叠而成,而自然界中无双晶存在的石英大多全部是由左形α-石英或右形α-石英组成。由此推测,尺度大多为纳米级的斜硅石微晶在α-石英中的出现可以视作一种结构缺陷,在一定程度上破坏了α-石英中质点规则排列所形成的长程有序结构,使以α-石英为主要成分的石英质玉石的结晶完整程度降低,从而使体现这种完整程度的结晶度指数下降。因此,图 4中能够表征斜硅石相对含量的I502/I465比值越高,其石英质玉石的结晶度指数越低。
石英质玉石结晶度指数的变化也可以通过X射线衍射图谱直观地表现出来。由于斜硅石相对含量的不同,石英质玉石中α-石英晶体结构的畸变程度也有所差异,这种结构畸变将导致在X射线衍射过程中,本应产生的衍射转化为不同程度的弥散散射。因此,图 3中斜硅石相对含量较高的样品,其衍射峰一般窄且尖锐(如样品TB1235),反之则宽且弥散(如样品ALS-MNS-1)。
不同石英质玉石中斜硅石相对含量的差异可能与其形成环境密切相关。目前,关于α-石英中斜硅石的形成仍存在较多争议,但多数认为斜硅石是在α-石英结晶的过程中形成的,而α-石英既可从流体中直接结晶,也可通过“opal-A→opal-CT/opal-C→隐晶质石英”这一序列相变形成[2, 5, 20, 27, 28]。在α-石英结晶过程中,受温度、介质的pH值、SiO2含量、杂质的种类和含量的变化等多种因素影响[1, 16, 19, 20],α-石英的结晶过程可能会发生改变,左形α-石英与右形α-石英沿(1011) 面方向的晶格面网沿一定方向交替堆叠排列,从而促成了斜硅石的结晶和生长。
3. 结论
在利用无损测试方法分析石英质玉石中斜硅石的相对含量方面,现有研究大多依靠拉曼光谱和红外光谱,而对石英结晶度的定量分析则主要借助X射线衍射技术。本文利用拉曼光谱、红外光谱和X射线衍射这三种宝玉石检测领域较为常用的测试方法,对我国4个代表性产地产出的不同颜色、不同结构类型的石英质玉石中斜硅石的相对含量差异进行了探讨,并在对比分析斜硅石相对含量与石英质玉石结晶度的过程中发现了两者之间的负相关关系,认为拉曼光谱中能够描述斜硅石相对含量变化的502 cm-1/465 cm-1两峰强度比值(X)与石英质玉石的结晶度指数(Y)的负相关关系大致符合线性方程Y=-0.36X+6.93(r=-0.94)。这一结论建立了斜硅石相对含量与石英质玉石结晶度两个不同研究方向的关联性,为判断石英质玉石的结晶程度提供了一种无损的检测方法,也为定性评价石英质玉石的品级、探讨不同产地石英质玉石的形成环境提供了新的借鉴依据。
值得指出的是,由于研究样品数量有限,还需更多的石英质玉石样品的测试数据,以期完善斜硅石的相对含量与石英质玉石结晶度之间的定量关系。
-
![]()
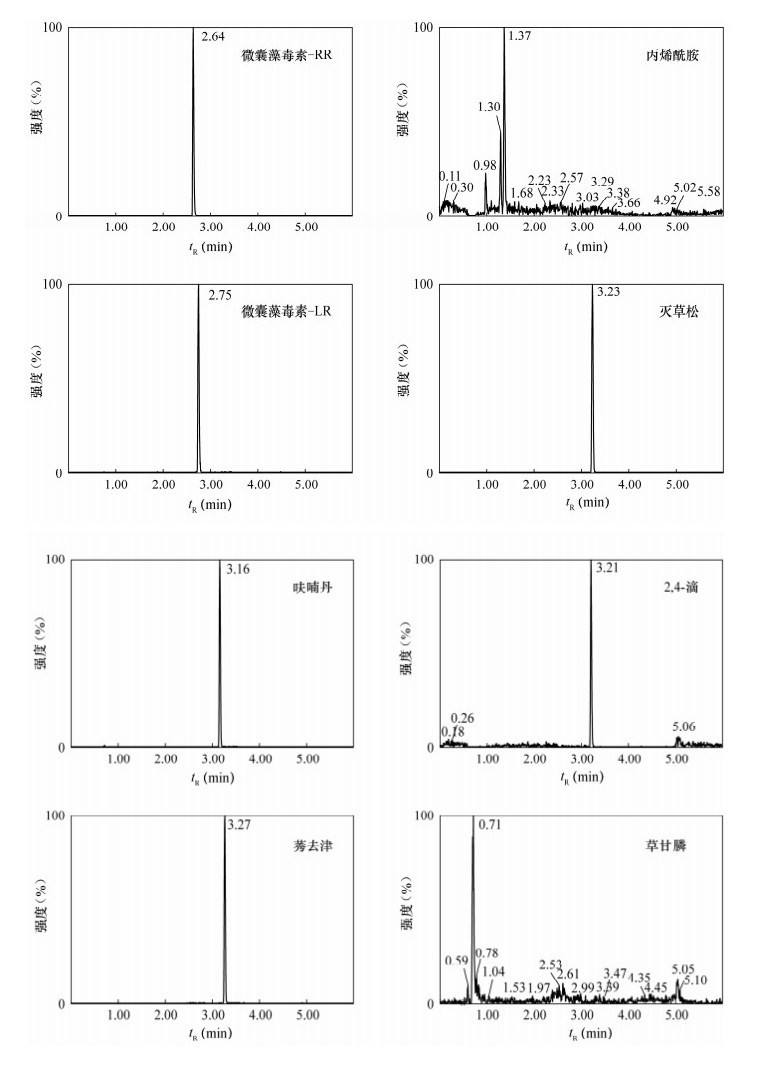
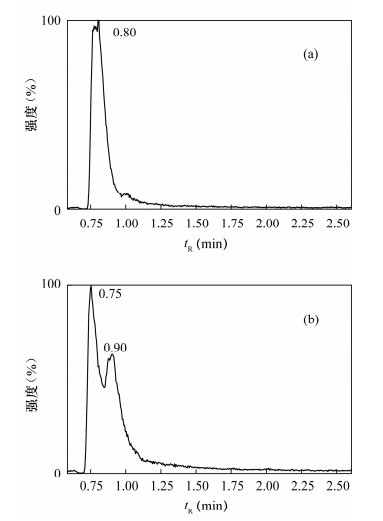
图 1 流动相不同时,草甘膦峰形比较色谱图
a—强洗脱相为甲醇的草甘膦色谱图;b—强洗脱相为乙腈的草甘膦色谱图。
Figure 1. Chromatograms with different elution solvents
![]()
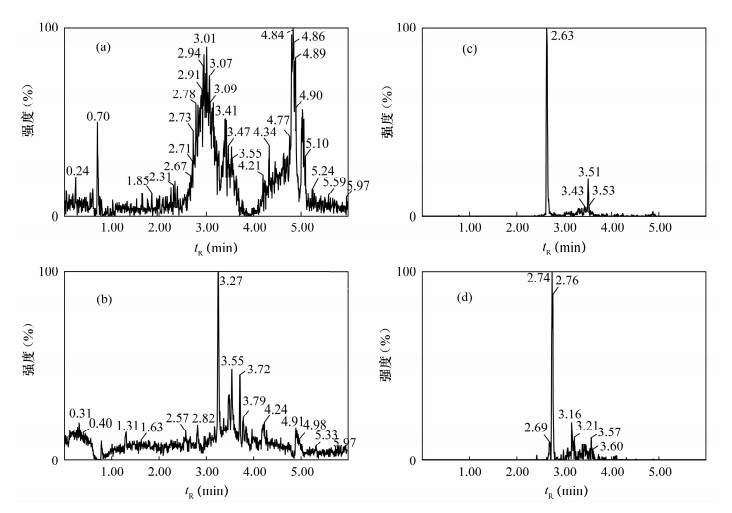
图 2 稀释20倍基质匹配混合标准溶液色谱图
Figure 2. Chromatograms of mixture standard solutions diluted 20 times by matrix matching
![]()
图 3 空白及阳性样品色谱图
a—正离子模式空白色谱图;b—负离子模式空白色谱图;c—微囊藻毒素-RR色谱图;d—微囊藻毒素-LR色谱图。
Figure 3. Chromatograms of blank and positive sample
表 2 8种有机污染物的监测离子对、碰撞能量、锥孔电压、离子化模式、检出限(S/N=3)及定量限(S/N=10)
Table 2 Qualitative ion pair, quantitative ion pair, collision energy, cone voltage, ionization mode, LOD(S/N=3) and LOQ (S/N=10) of the 8 organic pollutants
化合物 母离子
(m/z)定量离子
(m/z)定性离子
(m/z)碰撞能量
(eV)锥孔电压
(V)离子化模式 检出限
(μg/L)定量限
(μg/L)限量标准
(μg/L)草甘膦 168.0 150.0 142.0 10 30 ESI- 1.30 5.00 700 丙烯酰胺 72.0 55.0 44.0 8 35 ESI+ 0.12 0.30 0.5 2,4-滴 219.0 125.0 161.0 22 30 ESI- 0.02 0.07 30 灭草松 239.0 132.1 197.1 30 30 ESI- 0.03 0.10 300 莠去津 216.0 145.0 173.0 20 35 ESI+ 0.05 0.17 2 呋喃丹 222.0 122.9 165.1 15 30 ESI+ 0.03 0.10 7 微囊藻毒素-LR 498.5 135.2 213.2 30 30 ESI+ 0.02 0.07 1 微囊藻毒素-RR 520.1 282.2 282.2 18 30 ESI+ 0.02 0.07 1  下载: 导出CSV
下载: 导出CSV
表 1 8种有机污染物的保留时间、线性范围、相关系数
Table 1 The retention time, linear range, correlation coefficient of the 8 organic pollutants
化合物 保留时间
(min)线性范围
(μg/L)相关系数 混合标准液中的浓度
(μg/L)草甘膦 0.78 10~100 0.9955 500 丙烯酰胺 1.37 1~10 0.9973 50 2,4-滴 3.20 1~10 0.9982 50 灭草松 3.22 1~10 0.9932 50 莠去津 3.26 0.5~5 0.9991 25 呋喃丹 3.17 1~10 0.9955 50 微囊藻毒素-LR 2.76 1~10 0.9935 50 微囊藻毒素-RR 2.64 1~10 0.9998 50
下载: 导出CSV
表 3 8种有机污染物在自来水、地表水、地下水空白添加水平、回收率及精密度(n=6)
Table 3 The spiked levels, recovery and RSD (n=6) of the 8 organic pollutants in tap waters, surface waters and groundwaters
化合物 添加水平
(μg/L)自来水 地表水 地下水 高水平 中水平 低水平 高水平 中水平 低水平 高水平 中水平 低水平 高 中 低 回收率
(%)RSD
(%)回收率
(%)RSD
(%)回收率
(%)RSD
(%)回收率
(%)RSD
(%)回收率
(%)RSD
(%)回收率
(%)RSD
(%)回收率
(%)RSD
(%)回收率
(%)RSD
(%)回收率
(%)RSD
(%)草甘膦 100 20 10 97..2 5.2 97.8 3.5 96.5 2 95.3 2.3 94.2 4.3 103.7 1.8 98 3.2 102.1 6.5 96.3 4.1 丙烯酰胺 10 2 1 98.1 3.1 96.7 2.7 101.7 1.7 97.6 4.7 98.7 3.5 101.5 5.4 95.6 2 103.6 5.3 96.7 1.5 2,4-滴 10 2 1 99.1 2.1 99.5 2.4 99 2.7 98.4 5.2 103.2 7.8 97.2 6.3 96.3 3.4 99.8 7.4 99.6 4.2 灭草松 10 2 1 98.5 2.5 100.3 1.6 98.3 1.4 102.1 1.6 102.1 1.5 96 7 96 1.8 99.3 1.2 95 2.3 莠去津 5 1 0.5 99.3 1.3 100.5 0.8 99.6 3.2 99.1 1.4 99.5 6.4 95.3 2.4 97.7 1.4 99.7 1.6 98.3 3.6 呋喃丹 10 2 1 101.2 3.5 98.9 1.5 101.2 2.5 95.2 6.8 96.4 5.2 98.7 3.5 98.5 3.6 95.3 1.7 97.1 2.4 MCYST-LR 10 2 1 99.5 2.2 98.7 1.3 99.9 1.1 96.3 6.2 94.8 3.1 100 7.1 97.4 7.6 96.2 3.2 99.2 1.8 MCYST-RR 10 2 1 98.6 1.5 99.5 2.2 100.5 2.4 95.2 5.2 97.2 3.7 94.9 4.3 96.2 6.1 98.1 5.1 96.3 3.1
下载: 导出CSV
-
[1] 岳舜琳.世界卫生组织《饮水水质准则》--安全饮用水的标准[J].净水技术,2004(增刊):25-30. http://www.cnki.com.cn/Article/CJFDTOTAL-ZSJS2004S1015.htm [2] World Health Organization.Guidelines for Drinking-Water Quality (3th Edition)[S].
[3] GB 5749-2006,生活饮用水卫生标准[S] [4] GB 5750-2006,生活饮用水标准检验方法[S]. [5] 李会仙,吴丰昌,陈艳卿,张瑞卿,赵晓丽,廖海清,郭广慧.我国水质标准与外国水质标准/基准的对比分析[J].中国给水排水,2012,28(8):15-18. http://www.cnki.com.cn/Article/CJFDTOTAL-GSPS201208005.htm [6] 刘丽仪,陈小辉,唐励文.SPE-NPD气相色谱法测定水中莠去津和甲草胺除草剂[J].分析试验室,2007,26(增刊):163-164. http://www.cnki.com.cn/Article/CJFDTOTAL-FXSY2007S1053.htm [7] 张鑫,丁清波.气相色谱法测定水中呋喃丹[J].环境监测管理与技术, 2007,19(2):36-38. http://cpfd.cnki.com.cn/Article/CPFDTOTAL-CQYF201103001062.htm [8] 朱铭洪,马永建.测定水中丙烯酰胺方法的研究[J].江苏预防医学[J].2012, 23(1):65-66. http://www.cnki.com.cn/Article/CJFDTOTAL-IAOB201201015.htm [9] 陈锋,蔡少杰,李庆云,刘丽仪.高效液相色谱法测定水中酸性除草剂灭草松和2,4-滴[J].分析试验室,2008,27(增刊):286-288. [10] 薛刚,杨琳,龚清杰,闵浩.高效液相色谱法测定水中微囊藻毒素[J].中国给水排水, 2009,25(22):90-97. doi: 10.3321/j.issn:1000-4602.2009.22.025 [11] Amelin V G, Bol'shakov D S, Tretiakov A V. Determin-ation of glyphosate and aminomethylphosphonic acid in surface water and vegetable oil by capillary zone electrophoresis[J].Journal of Analytical Chemistry, 2012, 67(4):386-391. doi: 10.1134/S1061934812020037
[12] Mo W Q, Liu J M, Cheng M, Chao M.Determination of 2,4-D, bentazone and atrazine in water by high performance liquid chromatography[J].Journal of Environment and Health,2011,28(6):532-534.
[13] Cernoch I, Franek M,Diblikova I, Hilscherova K, Randak T, Ocelka T, Blaha L.Determination of atrazine in surface waters by combination of POCIS passive sampling and ELISA detection[J].Journal of Environment Monitoring, 2011,13(9):2582-2587. doi: 10.1039/c1em10112a
[14] Cong L M, Huang B F, Qi C, Cai Z X, Ren Y P.Determination of trace amount of microcystins in water sample using liquid chromotography coupled with triple quadrupole mass spectrometry[J].Analytica Chimica Acta, 2006, 569:157-168. doi: 10.1016/j.aca.2006.03.052
[15] Xu W, Chen Q, Zhang T, Cai Z X, Jia X F, Xie Q, Ren Y P.Development and application of ultra performance liquid chromatography-electrospray ionization tandam triple quadrople mass spectrometry for determination of seven microcystins in water samples[J].Analytica Chimica Acta, 2008,626:28-36. doi: 10.1016/j.aca.2008.07.040
[16] 张明,唐访良,陈峰,徐建芬,张丽娜.固相萃取-超高效液相色谱-电喷雾串联质谱法同时测定地表水中9种微囊藻毒素[J].色谱,2012, 30(1):51-55. http://www.cnki.com.cn/Article/CJFDTOTAL-SPZZ201201011.htm [17] 张春燕,赵兴茹,郑学忠.在线固相萃取超高效液相色谱串联质谱法测定水中微囊藻毒素[J].环境化学[J].2012,31(10):1663-1665. http://www.cnki.com.cn/Article/CJFDTOTAL-ZSJS201303012.htm [18] Renata R, Michele L E.Liquid chromatography with post-column reagent addition of ammonia in methanol coupled to negative ion electrospray ionization tandem mass spectrometry for determination of phenoxyacid herbicides and their degradation products in surface water[J].Analysis Chemistry Insights,2010, 5:1-14.
[19] 郑和辉,李洁,魏建荣,吴大南,王萍.液相色谱串联质谱法直接进样测定水中的莠去津,呋喃丹,灭草松[J].卫生研究, 2009,38(3):302-303. http://www.cnki.com.cn/Article/CJFDTOTAL-GSPS201518029.htm -
期刊类型引用(19)
1. 姜冰,张德明,刘阳. 大气降尘对土壤主要环境元素的累积影响及评价——以山东省高密市为例. 物探与化探. 2024(01): 228-236 .  百度学术
百度学术
2. 廖琦,肖粤新,杨鹏至,卢欣,毛雄. 汨罗市白塘镇-桃林寺镇土壤镉元素分布特征及成因分析. 四川地质学报. 2024(02): 294-298+305 . 百度学术
3. 张喜亮,闵宁,程成,陈超,李田田,蒋家沁. 皖北某电厂周边土壤重金属污染现状及评价分析. 河南科技. 2023(02): 79-82 . 百度学术
4. 韩跃明 ,刘思婕 ,王婉玥 . ICP测定玉米地中5种重金属含量方法研究. 高原农业. 2022(03): 220-224 . 百度学术
5. 刘进,潘月鹏,师华定. 华北地区农田土壤镉来源及大气沉降的贡献. 农业环境科学学报. 2022(08): 1698-1708 . 百度学术
6. 胡冠钊,乔少博,王驹,陈风敏,殷炜昭,宫艳艳. 广州市南沙区高新沙水库大气干湿沉降重金属含量、沉降通量特征分析及其对水库水质的影响. 环境科学学报. 2022(10): 120-128 . 百度学术
7. 张桂芹,谭路遥,张怀成,董磊磊,杜云龙,朱丽. 济南城市主干道降尘重金属污染特征及生态风险评价. 生态环境学报. 2020(01): 156-164 . 百度学术
8. 刘景霞,李赟. 生活垃圾焚烧发电厂烟尘中重金属沉降对土壤环境的影响研究. 绿色科技. 2020(10): 64-65+71 . 百度学术
9. 庞绪贵,代杰瑞,喻超,任天龙,刘华峰,张华平,曹洪松,曾宪东,任文凯,王增辉,赵西强,王存龙,王红晋,董健. 山东省17市土壤地球化学基准值. 山东国土资源. 2019(01): 36-45 . 百度学术
10. 庞绪贵,代杰瑞,陈磊,刘华峰,喻超,韩鎏,任天龙,胡雪平,王红晋,王增辉,赵西强,曾宪东,任文凯,王存龙. 山东省17市土壤地球化学背景值. 山东国土资源. 2019(01): 46-56 . 百度学术
11. 蔡敬怡,谭科艳,路国慧,殷效彩,郑宇,邵鹏威,王竞,杨永亮. 贵州万山废弃矿区小流域系统沉积物及悬浮物重金属的空间分布特征. 岩矿测试. 2019(03): 305-315 . 本站查看
12. 潘光,李少洛,朱丽,孙友敏,张桂芹. 济南市降尘通量时空分布特征研究. 生态环境学报. 2019(09): 1802-1809 . 百度学术
13. 王佳,刘斌,肖柏林,李余杰,张田硕,吴璜,张玉婷. 重庆主城区空气降尘中重金属的特点及其在表层土壤中的累积量研究. 土壤. 2019(06): 1160-1167 . 百度学术
14. 邱海鸥,包娅琪,孔贝贝. 原子光谱分析的研究进展及应用现状. 分析试验室. 2018(01): 108-124 . 百度学术
15. 庞绪贵,宋娟娟,代杰瑞,喻超,张华平,陈磊,任天龙,王红晋. 日照市土壤地球化学元素分布规律及成因探讨. 山东国土资源. 2018(04): 43-49 . 百度学术
16. 李娟,谭洪涛,周鸿,谢慧英,辜芸. 自动测汞仪直接测定PM_(2.5)中的汞. 实验与检验医学. 2018(05): 669-670 . 百度学术
17. 杨晓天,刘洋. 湖北省武汉市大气降尘的化学特征的光谱图像特征识别. 科技通报. 2017(04): 19-23 . 百度学术
18. 南茂才. 涟源地区大气干湿沉降通量研究及地球化学等级划分. 城市地理. 2016(14): 97-99 . 百度学术
19. 程秀花,王海蓉,黎卫亮,王鹏. 电感耦合等离子体质谱法测定硒时多元素干扰的碰撞/反应研究及其在地质样品中的应用. 冶金分析. 2015(12): 5-9 . 百度学术
其他类型引用(17)




计量
- 文章访问数: 1034
- HTML全文浏览量: 327
- PDF下载量: 14
- 被引次数: 36
 京公网安备 11010202008159号
京公网安备 11010202008159号