
Accurate Measurement of Argon Isotope Composition of Air by Argus Multi-collector Noble Gas Mass Spectrometer
-
摘要:
稀有气体质谱仪准确测量氩同位素组成是Ar-Ar法高精度定年的前提,目前测量氩同位素主要应用单接收或多接收质谱仪,其中多接收稀有气体质谱仪在数据准确性和重现性等方面具备优势。本文研究了Argus多接收稀有气体质谱仪应用于测量Ar同位素过程中一些主要因素对测量结果准确度和重现性的影响情况。结果表明,整套系统在静态模式下不同时间段的本底值极低,不影响测定;仪器电子倍增器的接收效率优于99.67%,可显著提高Ar低含量样品测量精度,当40Ar信号强度低于0.5 V时,用电子倍增器测量40Ar/36Ar组成的标准偏差仅为0.11%,而用法拉第杯测量40Ar/36Ar组成的标准偏差为0.53%;仪器的质量歧视效应可通过多次循环测量并采用指数定律获得稳定的质量歧视校正因子(此值相对标准偏差为0.0434%),实现对Ar同位素组成的准确校正。本文以测量空气中的氩同位素组成为例,证明了Argus多接收稀有气体质谱仪的测试效率比单个接收器跳峰方式的测试效率高,测试结果更精确,因此适合年轻样品或含钾量极低的样品的Ar-Ar高精度定年工作。
Abstract:Accurate measurement of Argon isotopes is the premise of Ar-Ar dating with high precision. Two main kinds of instruments for Ar isotope measurement are Single-collector and Multi-collector Mass Spectrometers, among which the Multi-collector Noble Gas Mass Spectrometer can produce data with better accuracy and reproducibility. Some main factors affecting the accuracy and reproducibility of the results during the measurement of the Ar isotopes by the Argus Multi-collector Noble Gas Mass Spectrometer are discussed in this paper. Results show that the background of the whole system in static mode is very low and has little effect on the measurement of Ar isotope composition. The efficiency of the multiplier of the instrument is better than 99.67%, which can improve the measurement precision of low Ar samples. When the signal of 40Ar is less than 0.5 V, the standard error is only 0.11% for the 40Ar/36Ar ratios measured by the CDD, while the standard error is 0.53% when the ratios are measured using a Faraday cup. The mass discrimination effect of the instrument could be calibrated with the logarithmic law through the repeated measurement of argon isotopes in air and the relative standard error of the mass discrimination is only 0.0434%. Using the example of the determination of argon isotopes in air, it can be demonstrated that the efficiency of the measurement by Argus Multi-collector Mass Spectrometer is higher than that of the Single-collector Mass Spectrometer with higher precision and accuracy. Therefore, the Multi-collector Mass Spectrometer is highly suitable for the high-precision Ar-Ar dating of young and potasium-poor samples.
-
我国铝土矿资源以一水硬铝石为主,主要集中在山西、河南、广西、贵州四省[1]。它具有较强的化学稳定性,常常含有少量刚玉(Al2O3),属于比较难处理的样品。铝土矿的基本分析项目共5项:二氧化硅、三氧化二铝、三氧化二铁、二氧化钛、烧失量[2]。目前测定铝土矿中化学成分的常规方法有重量法[3-4]、滴定法[4-6]、分光光度法[4, 7-8]等,但这些方法测定周期长,过程复杂,工作强度大,技术水平要求高,不能多元素同时测定。近年来,电感耦合等离子体发射光谱法(ICP-AES)因其灵敏度高、准确性好、测试范围宽、多元素同时测定等优点被广泛应用于合金和铝土矿分析[9-11]。与之配套的铝土矿样品分解一般有碱熔法和混合酸溶解法[12-13],常用的四酸溶解法对不含刚玉的铝土矿能取得较好的效果,但对高铝及含刚玉的样品分解不完全且还无法同时测定硅。用偏硼酸锂在铂坩埚中熔融铝土矿,虽然样品分解完全,但熔融温度高,造成提取困难[14]。一水硬铝石往往与刚玉共生,单独采用氢氧化钠或氢氧化钾熔融,对含刚玉的铝土矿分解不完全。
本文采用制作成本较低的镍坩埚,对含少量刚玉的铝土矿样品用氢氧化钠+过氧化钠熔融,所得熔融体用盐酸提取后采用ICP-AES测定硅、铝、铁、钛的含量,解决了此类铝土矿分解不完全的问题,实现了准确测定。
1. 实验部分
1.1 仪器及工作条件
Prodigy XP电感耦合等离子体发射光谱仪(美国Leeman公司),双铂网雾化器。
仪器工作参数为:射频功率1200 W,雾化气压力0.33 MPa,冷却气流量20 L/min,辅助气流量0.3 L/min,重复测定次数为2次,样品提升速度1.4 mL/min,冲洗时间40 s,积分时间15 s,观测方式:垂直。氩气纯度 > 99.995%。
1.2 标准物质及主要试剂
铝土矿国家标准物质(GBW 07177、GBW 07178、GBW 07180)。
土壤国家标准物质(GBW 07405):作为二氧化硅标准曲线的高浓度点。
铁元素标准储备溶液:1 mg/mL(国家钢铁材料测试中心)。
氢氧化钠(分析纯,天津市化学试剂三厂),过氧化钠(分析纯,天津大沽化工股份有限公司)。
盐酸(分析纯,洛阳昊华化学试剂有限公司)。
实验用水为二次去离子水。
1.3 实验方法
强碱性熔剂(过氧化钠、氢氧化钠或氢氧化钾)是分解铝土矿最有效的熔剂。对于含有刚玉的铝土矿,仅用氢氧化钠仅用或氢氧化钾不能完全熔融,须加入一定量的过氧化钠。本文试验了氢氧化钠+过氧化钠、氢氧化钠两类熔剂,考察对含有刚玉的铝土矿样品熔解效果。
方法1:氢氧化钠+过氧化钠熔融。称取0.5000 g(精确至0.0002 g)试样置于30 mL镍坩埚中,滴加几滴无水乙醇润湿样品,覆盖约3.0 g氢氧化钠,置于低温马弗炉中,升温至550℃左右。在此温度下保温5 min,取出稍冷,再加入1.0~1.2 g过氧化钠,重新放入马弗炉内升温至700℃,在此温度下保温10 min,取出坩埚冷却后,放入250 mL烧杯中,滴加几滴无水乙醇,加50 mL热水浸取,尽快加入浓盐酸搅拌至反应停止,再加25 mL浓盐酸,加热至溶液澄清透明,取下冷却,将溶液转移至250 mL容量瓶中,用水稀释至刻度,摇匀,分取10 mL于100 mL容量瓶内,加10 mL盐酸,用水稀释至刻度,摇匀,用ICP-AES测定。同时做试剂空白试验。
方法2:氢氧化钠熔融。称取0.2500~0.5000 g(精确至0.0002 g)试样置于50 mL镍坩埚中,滴加几滴无水乙醇润湿样品,覆盖3.0~4.0 g氢氧化钠,置于低温马弗炉中,升温至700℃。在此温度下保温5~10 min,取出镍坩埚放入250 mL烧杯中,滴加几滴无水乙醇,加50 mL热水浸取,尽快加浓盐酸搅拌至反应停止,再加25 mL浓盐酸,加热至溶液澄清透明,取下冷却,将溶液转移至250 mL容量瓶中,用水稀释至刻度,摇匀,分取10 mL于100 mL容量瓶内,加入10 mL盐酸,用水稀释至刻度,摇匀,用ICP-AES测定。同时做试剂空白试验。
1.4 标准溶液配制
制备的空白试验溶液为空白,用铝土矿国家标准物质(GBW 07177、GBW 07178、GBW 07180、GBW 07405)按1.3节方法1制备的溶液为标准系列。铝土矿国家标准物质样品不含有高含量的铁,而实际的地质样品有很多铁含量高于15%的高铝铝土矿,因此分取10 mL空白溶液,加入适量的ρ(Fe)=1 mg/mL标准溶液,根据样品中铁的含量配制2个介质基本相同的人工标准溶液。具体浓度见表 1。
表 1 标准溶液Table 1. Standard solutions标准溶液 w/% SiO2 Al2O3 Fe2O3(Fe) TiO2 STD1(GBW 07177) 7.80 71.06 1.82 3.08 STD2(GBW 07178) 15.24 54.94 9.04 2.46 STD3(GBW 07180) 36.03 42.97 0.41 2.06 STD4(GBW 07405) 52.57 21.58 12.62 1.05 STD5(人工标准溶液) - - 15.00 - STD6(人工标准溶液) - - 25.00 - 注:为了测量结果便于计算,表中溶液浓度已换算为固体含量。 2. 结果与讨论
2.1 熔矿方式及坩埚材质的选择
本方法利用氢氧化钠和过氧化钠的强氧化性可以完全分解高铝铝土矿,特别是含有刚玉的一水硬铝石。
铝土矿熔融分解常用的坩埚材质有:镍、银、铂、热解石墨坩埚。因为铂(银)坩埚价格昂贵,采用过氧化钠熔融,如果温度超过550℃,铂(银)坩埚的腐蚀大幅上升。550℃熔融数分钟,过氧化钠对铂坩埚的腐蚀大于30 mg以上[15]。而热解石墨坩埚不耐强氧化性酸碱熔剂的侵蚀,易损。镍坩埚价格较低,熔融损失不是很大,可满足日常分析工作的应用。
2.2 氢氧化钠+过氧化钠与氢氧化钠熔融能力的对比
选用铝土矿国家标准物质GBW 07181、GBW 07182,这两种标准物质因Al含量高,采用氢氧化钠反复熔融分解不完全,酸化提取后杯底有残渣,经查找相关文献和岩矿鉴定为刚玉[16]。称取每个标准物质各两份,称样量为0.25 g,一份加入氢氧化钠+过氧化钠﹙方法1﹚,另一份加入氢氧化钠﹙方法2),熔融提取。氢氧化钠的熔融物经提取后,杯底有近似透明不溶残渣,而氢氧化钠+过氧化钠的熔融物经提取后,杯底无残渣,ICP-AES测定结果见表 2。
表 2 两种熔样方法比较Table 2. Comparison of analytical results with two dissolution methodsw/% 标准物质编号 熔样方法 SiO2 Al2O3 Fe2O3 TiO2 标准值 平均值 RE/% 标准值 平均值 RE/% 标准值 平均值 RE/% 标准值 平均值 RE/% GBW 07181 方法1 3.2 3.21 0.31 90.63 90.51 -0.13 1.31 1.32 0.76 3.8 3.77 -0.79 方法2 3.2 3.16 -1.25 80.36 -11.33 1.31 1.24 -5.34 3.8 3.65 -3.95 GBW 07182 方法1 19.44 19.26 -0.93 75.13 75.32 0.25 1.24 1.32 6.45 3.22 3.33 3.42 方法2 19.44 18.59 -4.37 66.3 -11.75 1.24 1.18 -4.84 3.22 3.05 -5.28 采用方法2熔样时Si、Al、Fe、Ti系统偏低,特别是Al明显偏低,测定值与标准值的相对误差(RE)大于10%。而方法1熔融的测定值无系统误差,相对误差为-0.93%~6.45%。可见用氢氧化钠熔融,即便熔剂用量为样品质量的12倍,依然无法完全熔解含少量刚玉的铝土矿样品,而采用氢氧化钠+过氧化钠的熔矿方法能够完全熔解高铝及含少量刚玉的铝土矿。
2.3 过氧化钠的加入量
对过氧化钠的加入量进行了实验。称取0.5000 g铝土矿标准物质GBW 07182样品5份,按1.3节实验方法1首先加入氢氧化钠后,置于马弗炉低温加热并熔融,取下稍冷,分别加0.5、0.8、1.0、1.2、1.5 g过氧化钠后,700℃熔融10 min。由表 3的测定结果可见,加入0.5、0.8 g过氧化钠提取酸化后的溶液的测定值比标准值明显偏低,溶液中有少量矿物残渣存在;加入1.0、1.2、1.5 g过氧化钠提取酸化后的溶液的测定值与标准值吻合,且溶液澄清透明无残渣颗粒存在。为了保证样品分解完全并使溶液盐分尽量少,本文选择加入1.0~1.2 g过氧化钠熔融样品,对部分高铝及含刚玉的铝土矿可适当增加过氧化钠的用量至1.5 g。
表 3 过氧化钠用量的选择Table 3. The selection of sodium peroxide amount过氧化钠加入量
m/gw/% SiO2 Al2O3 Fe2O3 TiO2 0.5 18.76 74.00 1.22 3.20 0.8 18.94 74.50 1.24 3.25 1.0 19.39 75.38 1.26 3.20 1.2 19.55 74.99 1.24 3.24 1.5 19.35 75.06 1.25 3.23 GBW 07182标准值 19.44 75.13 1.24 3.22 2.4 熔样温度
对一般的铝土矿在650℃下熔样5~10 min即可分解完全,且可减轻对坩埚的腐蚀。对含刚玉的铝土矿,用氢氧化钠+过氧化钠熔融时,由于过氧化钠的强氧化性和不稳定性,一般都是在较高温度下置于马弗炉中,以利用其较强的氧化性分解矿样。
本实验采取先加氢氧化钠在低温马弗炉熔融至550℃时,冷却后再加过氧化钠重新入炉熔融至700℃,结果表明650~700℃熔融10 min可保证试样熔解完全,温度大于700℃时对坩埚腐蚀较严重,溶液呈瓦蓝色。由标准物质GBW 07182的分析结果可见,测定值与标准值基本吻合(表 4)。因此确定加入一定量过氧化钠后在650~700℃保温10 min熔融样品。
表 4 熔样温度Table 4. Melting temperature of samples熔样温度
θ/℃w/% SiO2 Al2O3 Fe2O3 TiO2 600 18.57 71.49 1.18 3.00 650 19.35 74.65 1.23 3.12 700 19.40 75.08 1.25 3.23 730 19.55 75.10 1.18 3.24 GBW 07182标准值 19.44 75.13 1.24 3.22 2.5 分析谱线的选择及方法检出限
每个元素选择3~4条谱线,经过对试样及标准样品溶液的多次扫描,比较了图谱、背景轮廓及测定过程的稳定性和结果的准确性。在仪器最佳条件下对试剂空白溶液连续测定11次,以测定结果的10倍标准偏差乘以稀释因子(稀释因子为5000)计算方法检出限。选定谱线、背景校正位置及方法检出限见表 5。
表 5 元素分析谱线、背景校正位置和方法检出限Table 5. Spectral lines and detection limits of the method元素 分析谱线λ/nm 背景校正 方法检出限/% SiO2 212.412 右3 左3 0.063 251.611 右3 左3 0.011 Al2O3 308.215 右3 左3 0.033 396.152 右3 左3 0.024 Fe2O3 239.563 右3 左2 0.024 248.814 右4 左3 0.051 TiO2 334.941 右2 左1 0.0025 337.280 右3 左3 0.013 2.6 共存离子影响及基体干扰消除
由于采用镍坩埚,氢氧化钠+过氧化钠熔样,试液中有大量的镍盐和钠盐基体,经理论计算熔融加3.0 g氢氧化钠、1.0 g过氧化钠提取酸化定容,溶液中Na含量高达9.3 mg/mL,远远高出ICP-AES所限制的1 mg/mL的含盐量。大量的Na使ICP-AES的雾化器极易堵塞,为避免钠盐对分析结果的严重影响,氢氧化钠和过氧化钠保持定量加入后分取将母液大比例稀释。上机测定时采用耐高盐的双铂网雾化器,用标准物质(GBW 07177、GBW 0 7178、GBW 07180、GBW 07405)制备溶液绘制标准曲线,可减轻钠盐和基体对分析的影响,而用标准溶液配制的混合标准溶液,没有加入钠盐和其他基体,测定值波动较大。
镍坩埚熔融样品,溶液中也含有较高的镍盐,因此选择Ni含量为1 mg/mL的单元素标准溶液,分别对待测元素Ti、Al、Fe、Si进行干扰试验,从待测元素与Ni的谱图叠加来看,所选Ti、Al的谱线不受Ni的干扰,Fe、Si谱线干扰程度轻微。样品溶液分取稀释后Ni含量低,干扰可以忽略不计。对待测元素选择两条谱线,如果分析结果相差较大,对较高含量的元素以灵敏度低的谱线结果为准,对较低含量的元素以灵敏度高的结果为准,并从谱图叠加中查找可能的干扰元素。
2.7 方法准确度和精密度
选用铝土矿国家标准物质(GBW 07179、GBW 07182)独立处理并测定11次,计算其相对误差和相对标准偏差,结果见表 6,测定值与标准值相符,准确度小于3%,方法精密度(RSD)小于4%。
表 6 方法准确度和精密度Table 6. Accuracy and precision tests of the method标准物质编号 元素 w/% 相对误差
RE/%精密度
RSD/%平均值 标准值 GBW 07179 SiO2 16.56 16.62±0.09 -0.36 0.5 Al2O3 63.13 63.17±0.25 -0.06 0.2 Fe2O3 0.69 0.68±0.03 1.47 3.0 TiO2 3.29 3.28±0.03 0.30 0.5 GBW 07182 SiO2 19.48 19.44±0.40 0.21 0.5 Al2O3 75.14 75.13±0.45 0.01 0.2 Fe2O3 1.24 1.24±0.04 0.00 2.9 TiO2 3.23 3.22±0.03 0.31 1.2 2.8 实际样品分析和结果对照
选用未知样品1#~3#,并将本法的测定结果与其他方法(重量法、分光光度法、滴定法)测定结果进行对比。从表 7可以看出,本法测定结果与其他分析方法所测结果无显著性差异。
表 7 实际样品分析Table 7. Analytical results for actual samplesw/% 元素 1#样品 2#样品 3#样品 本法 其他方法 本法 其他方法 本法 其他方法 SiO2 28.49 28.32 8.45 8.38 23.78 23.95 Al2O3 51.27 51.11 71.45 71.60 26.01 25.90 Fe2O3 1.60 1.61 3.01 2.99 25.12 25.09 TiO2 2.53 2.55 0.80 0.83 1.24 1.22 注:表中其他方法为,SiO2为重量法测定,Al2O3为滴定法测定,Fe2O3、TiO2为分光光度法测定。 3. 结语
本文采用镍坩埚,氢氧化钠+过氧化钠熔融铝土矿样品,解决了四酸溶样或单纯氢氧化钠熔融含刚玉的铝土矿分解不完全的问题。利用铝土矿标准物质制备标准溶液,消除了样品基体、溶液介质等因素对硅、铝、铁、钛测定的干扰。本方法分解样品完全,样品处理过程简单,易于掌握。经国家标准物质和大量实际样品验证,测定结果具有良好的准确度和重现性,分析效率高,适用于高铝及含少量刚玉的铝土矿样品分析。
-
![]()
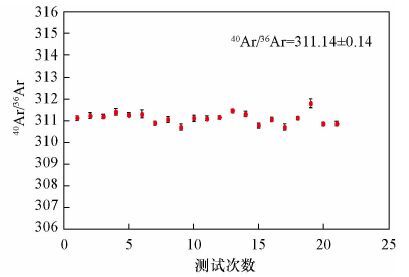
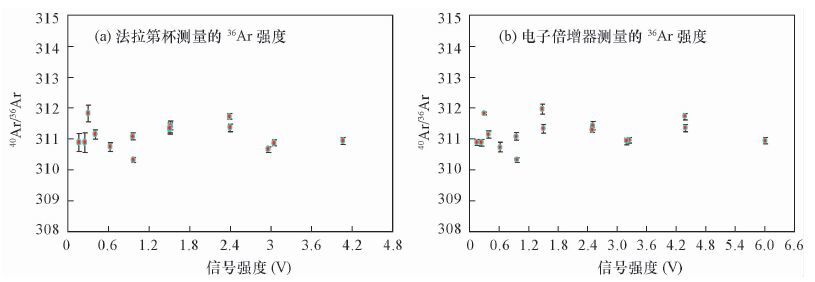
图 1 不同Ar强度下40Ar/36Ar测量值
Figure 1. Measurement of 40Ar/36Ar with different Ar signals (36Ar on Faraday Cup and CDD,respectively)
![]()
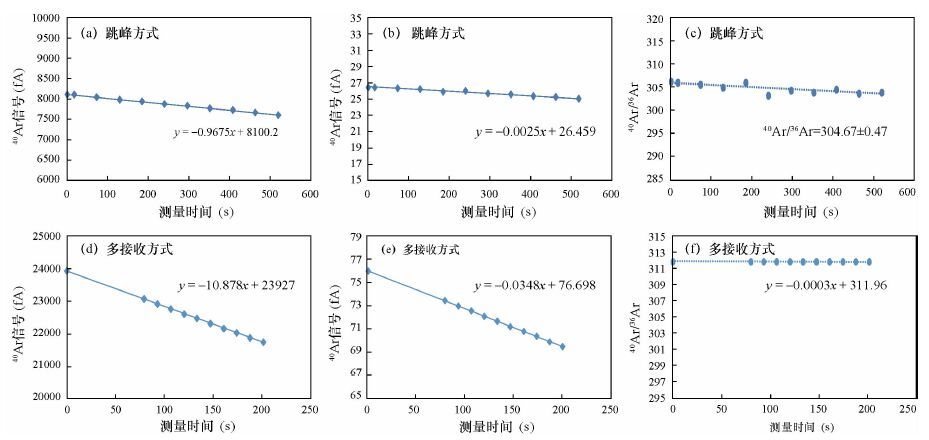
图 3 Argus Ⅵ 稀有气体质谱仪跳峰方式(a、b、c)和多接收方式(d、e、f)测量空气氩同位素组成
Figure 3. Measurement of Ar isotopes in air by Argus Ⅵ with (a,b,c) jump mode and (d,e,f) multi-collect mode
表 1 法拉第杯之间增益系数确定
Table 1 The determination of the gain between the Faraday cups
法拉第杯名称 UFC 补偿(fA) 增益系数 校正因子 H2 -13.9771985 0.0001179 1 H1 -0.7418739 0.0020749 1 AX -6.7486484 0.0019324 1 L1 -3.354546 0.0010854 1 L2 -1.6338639 0.0017126 1  下载: 导出CSV
下载: 导出CSV
表 2 电子倍增器的接收效率
Table 2 The receiving efficiency of the CDD SEM
循环次数 中心杯测量质量数:43.288 电子倍增器测量质量数:39.962 第一个法拉第杯测量质量数:39.962 中心杯测量质量数:38.962 电子倍增器的接收效率(%) 1 -0.147734 56.513159 56.635510 -0.154347 99.78 2 -0.131512 56.824269 56.905517 -0.099949 99.86 3 -0.064739 57.069140 57.321538 -0.099247 99.56 4 -0.095934 57.380536 57.502257 -0.066231 99.79 5 -0.149298 57.602783 57.887258 -0.068898 99.51 6 -0.160772 57.960536 57.994782 -0.037516 99.94 7 -0.009275 58.261207 58.384115 -0.082337 99.79 8 -0.093395 58.524347 58.599753 -0.165132 99.87 9 -0.114019 58.863647 58.937859 -0.179805 99.871 10 -0.119440 59.107328 59.381736 -0.110706 99.54 11 -0.151464 59.489227 59.584644 -0.086769 99.84 12 -0.115166 59.744958 59.899836 -0.116371 99.74 注:法拉第杯和离子倍增器信号强度均已扣除信号噪音值。AX代表中心法拉第杯,CCD SEM代表电子倍增器,H1代表第一个法拉第杯。
下载: 导出CSV
-
[1] Turrin B D,Swisher C C,Dieno A L.Mass Discrimination Monitor and Intercalibration of Dual Collectors in Noble Gas Mass Spectrometer Systems[J].Geochemistry,Geophysics,Geosystems,2010,11:doi: 10.1029/2009GC003013.
[2] Mark D F,Stuart F M,de Podesta M.New High Precision Measurement of the Isotopic Composition of Atmospheric Argon[J].Geochimica et Cosmochimica Acta,2011,75:7494-7501.
[3] Renne P R,Sharp W D,Dieno A L,et al.The Isotopic Composition of Atmospheric Argon and Ar/Ar Geochronology:Time for a Change?[J].Quarternary Geochronology,2009,4(4):288-298.
[4] Kim J,Jeon S.40Ar/39 Ar Age Determination Using Argus Ⅵ Multiple-collector Noble Gas Mass Spectrometer:Performance and Its Application to Geosciences[J].Journal of Analytical Science and Technology,2015,6:4-11.
[5] McDougall I,Harrison T M.Geochronology and Thermo-chronology by the 40Ar/39 Ar Method[M].Oxford:Oxford University Press,1999:89-90.
[6] Alexsander E C,Ozima M.Terrestrial Rare Gas[M].Tokyo:Center for Academic Publications,1978.
[7] Mark D F,Barfod D B,Stuart F M,et al.The Argus Multicollector Noble Gas Mass Spectrometer:Performance for Ar/Ar Geochronology[J].Geochemistry,Geophysics,Geosystem,2009,10(2):1-9.
[8] 李军杰,李剑,刘汉彬,等.Helix SFT惰性气体质谱仪分析矿物包裹体中氦同位素组成[J].地质学报,2015,89(10):1826-1831. Li J J,Li J,Liu H B,et al.The Analysis of Helium Isotope of Inclusions in Minernal Using Helix SFT Noble Gas Mass Spectrometer[J].Acta Geologica Sinica,2015,89(10):1826-1831.
[9] Eiler J M,Clog M, Magyar P,et al.A High-resolution Gas-source Isotope Ratio Mass Spectrometer[J].International Journal of Mass Spectrometry[J].http://dx.doi.org/10.1016/j.ijms.2012.10.014.2012.
[10] Turrin B D,Swisher C C,Deino A L.Mass Discrimination Monitoring and Intercalibration of Dual Collectors in Noble Gas Mass Spectrometer Systems[J].Geochemistry,Geophysics,Geosystem,2010,11.doi: 10.1029/2009GC003013.
[11] Burnard P G,Farley K A.Calibration of Pressure-dependent Sensitivity and Discrimination in Nier-type Noble Gas Ion Sources[J].Geochemistry,Geophysics,Geosystem,2000,1:33-38.
[12] Baur H A.Noble-gas Mass Spectrometer Compressor Source with Two Orders of Magnitude Improvement in Sensitivity[J].EOS,1999,80:1118.
[13] Reynolds J H.High Sensitivity Mass Spectrometer for Noble Gas Analysis[J].Review of Scientific Instrument,1956,27(11):928-934.
[14] Ryuji O,Tomoki N,Nobuo T,et al.Noble Gases in Ureilites Released by Crushing[J].Meteoritics and Planetary Science,2003,5:767-781.
[15] Ozima M,Podosek F A.Noble Gas Geochemistry[M].Cambridge:Cambridge University Press,2002:286.
[16] Baski A K,Archibald D A,Farrar E.Intercalibration of 40Ar/39 Ar Dating Standards[J].Chemical Geology,1996,129:307-324.
[17] Rudenhauer F G.Gas Scattering as a Limit to Partial-pressure Sensitivity[J].Vacuum Science Technology,1972,9:215.
[18] Lee J Y,Marti K,Severinghaus K,et al.A Redeter-mination of the Isotopic Abundances of Atmospheric Ar[J].Geochimica et Cosmochimica Acta,2006,70:4507-4512.
[19] Holst B,Buckland J R,Allison W.Spatial Mapping in the Electron Impact Ion Source of a Residual Gas Analyser[J].Vacuum,1999,53:207-210.
[20] Nier A O.A Redetermination of the Relative Abundances of the Isotopes of Carbon,Nitrogen,Oxygen,Argon and Potassium[J].Physics Review,1950,77:789-793.
[21] Young E D,Galy A,Nagahara H.Kinetc and Equilibrium Mass-dependent Isotope Fractionation Laws in Nature and Their Geochemical and Cosmochemical Significance[J].Geochmica et Cosmochimica Acta,2002,66(6):1095-1104.
[22] Valkiers S,Vendelbo D,Berglund M,et al.Preparation of Argon Primary Measurement Standards for the Calibration of Ion Current Ratios Measured in Argon[J].International Journal of Mass Spectrometry,2010,291:41-47.
计量
- 文章访问数: 1647
- HTML全文浏览量: 278
- PDF下载量: 14
 京公网安备 11010202008159号
京公网安备 11010202008159号