
Research Progress on Sample Preparation Methods and Analytical Techniques for Nickel Laterite
-
摘要: 随着常规镍来源的硫化镍矿资源的日益枯竭, 可直接生产氧化镍、镍锍和镍铁等产品的红土镍矿倍受关注.对于红土镍矿中主量、次量、痕量元素的检测, 相同的检测项目存在多种测试方法, 且部分相同原理的测试方法存在细节上的差异, 使得检测者选择合适的检测方法变得困难.本文综述了近年来红土镍矿中24种元素测定的样品前处理方式及分析技术研究进展.样品前处理方式依据目标元素及后续的分析方法进行选择, 其中酸溶法和碱熔法用途最广.酸溶法引入的盐分少, 操作简单, 但是分解过程中易导致挥发元素As、Sb、Bi、Hg的损失, Cr易随高氯酸冒烟损失.碱熔法分解能力强, 适合分析Cr、Si、全铁等项目, 但会引入大量的盐类和因坩埚材料损耗而带入其他杂质, 给后续分析带来困难.红土镍矿的分析技术依据实验室条件及目标元素的性质和浓度进行选择.电感耦合等离子体发射光谱法(ICP-AES)是主量、次量元素的主要分析方法, 适合于分析含量为10-5~30%级别的金属元素; X射线荧光光谱法主要用于分析含量为10-3~1级别的元素, 尤其适合于测定Al、Si、Ti、V和P, 由于该方法的准确性依赖于一套高质量的标准样品, 故更适合炉前检测或检测大批红土镍矿样品.电感耦合等离子体质谱法(ICP-MS)最适合于分析10-4含量以下的重元素, 特别是稀土和贵金属元素.原子吸收光谱法(AAS)适合于分析10-4~10-2级别的Ca、Mg、Ni、Co、Zn、Cr、Mn等低沸点、易原子化元素.分光光度法主要用于分析Ni和P.原子荧光光谱法(AFS)主要用于分析As、Bi、Sb等易形成气态氢化物的元素.容量法主要用于分析Al、Fe、Mg和SiO2等主含量元素.尽管AAS、分光光度法、AFS法和容量法检测周期长, 但所用仪器为实验室常规配置, 可满足缺乏相应大型仪器实验室的日常检测.本文认为, 针对各种检测方法的适用性及存在问题, 应从开发微波消解法、固体进样直接测汞法、ICP-MS法以及Cr与其他元素同时分析的快速分析方法等方面开展研究, 建立灵敏、准确的检测方法, 从而更好地服务于红土镍矿的贸易、检验和综合利用.
-
关键词:
- 红土镍矿 /
- 样品前处理技术 /
- 酸溶法 /
- 碱熔法 /
- 电感耦合等离子体发射光谱法 /
- X射线荧光光谱法 /
- 电感耦合等离子体质谱法 /
- 原子吸收光谱法 /
- 原子荧光光谱法 /
- 容量法
Abstract: With the depletion of common sulphide nickel resources, nickel laterite is becoming more attractive as nickel oxide, nickel matte and nickel-iron, amongst others, can be produced directly. For the determination of major, minor and trace elements in laterite nickel ore, many kinds of measurement methods for a given element and many differences of details between detection methods based on the same principle are available, making it difficult to select the appropriate method. Recent research in the pretreatment method and technology of the detection of a number of elements (Al, As, Bi, C, Ca, Cd, Cl, Co, Cr, Cu, F, Fe, Hg, Mg, Mn, Ni, P, Pb, S, Sb, Sc, Si, Ti, Zn) in laterite nickel is reviewed in this paper. The proper pretreatment methods were selected according to the target elements and detection method. Acid dissolution and alkali fusion as the pretreatment methods are most widely used now. Although acid dissolution introduces less salt and operates more simply, the volatile elements such as As, Sb, Bi and Hg are often lost in the decomposition process, while Cr is frequently lost by smoking with perchloric acid. Alkali fusion has better decomposition ability for Cr, Si and total iron, while the subsequent analysis is difficult, because impurities as well as many other salts are introduced due to the loss of crucible material and the melting process. The determination techniques of nickel laterite are chosen on the basis of character and concentration of target elements and the equipment in the laboratory. Inductively Coupled Plasma-Atomic Emission Spectrometry (ICP-AES), which is suitable for the determination of elements content from 10-5 to 30%, is the main analysis method of major, minor and trace elements. X-ray Fluorescence Spectrometry (XRF) is used to analysis elements content of 10-3-1 in samples, especially for the detection of Al, Si, Ti, V and P. This method whose accuracy relies on a set of high quality standard sample is suitable for spot detection and determination of the large numbers of laterite nickel ore samples. Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) is appropriate for determining heavy elements with the content of less than 10-4 in samples, especially for the rare earth and noble metal elements. Atomic Absorption Spectrometry (AAS) is adopted in the determination of the low boiling point elements with the content level ranging from 10-4 to 10-2 which are liable to atomization, such as Ca, Mg, Ni, Co, Zn, Cr and Mn. Spectrophotometry is mainly used for detecting the Ni and P, while Atomic Fluorescence Spectrometry (AFS) for the measurement of elements which were easily to form gaseous hydride such as As, Bi and Sb. Some main content elements, like Al, Fe, Mg and SiO2, are analyzed by Volumetry. Although the detection period is a little longer, AAS, Spectrophotometry, AFS and Volumetry which just need common equipment meet the daily determination in the laboratory which lacks large scale equipment. In order to solve the applicability and other existing problems, it is necessary to study the microwave digestion method, direct mercury analysis method, ICP-MS method, and rapid determination of Cr and other elements. The progress in establishing appropriate detection methods is certain to serve the trade, analysis and comprehensive utilization of laterite nickel ore. -
氨基甲酸酯和三唑类农药极性较强、热稳定性差,是目前广泛使用的广谱除草剂和高效杀菌剂、植物调节剂。然而,有证据表明氨基甲酸酯和三唑类杀菌剂农药对人体的内分泌、脊椎动物健康存在潜在毒性风险,特别是部分氨基甲酸酯农药的降解产物比原药具有更大毒性和持久性[1, 2]。土壤是农药残留的储存库,加速溶剂萃取(ASE)和超声提取(UE)是土壤样品中农药提取的主要方法[3, 4]。QuEChERS方法是2003年Anastassiades等[5]提出的一种针对蔬菜、水果样品中农药多残留检测的一种快速分析方法,它主要采用极性乙腈缓冲溶液匀浆提取样品,样品提取液经离心分离、分散固相萃取净化,离心后直接测定[6, 7, 8, 9]。迄今QuEChERS方法已在生物、植物样品中应用较为广泛[10, 11],但在土壤样品中应用很少,这是因为土壤对一般污染物有强烈吸附力,造成待测污染物回收率偏低。本文采用ASE、UE尝试提取土壤样品中的痕量氨基甲酸酯和三唑类农药,但其回收率偏低,这与ASE、UE属于热抽提方法、不适用于热稳定性差的目标物分析有关。在此基础上利用QuEChERS方法常温提取的特点,对提取溶剂和提取器皿进行了优化和改进,结合液相色谱-电喷雾串联质谱(LC-MS/MS)进行检测,基体加标回收率有所提高,此方法适用于土壤中一些极性较强、热稳定较差的有机污染物的分析。
1. 实验部分
1.1 仪器
美国Agilent公司1200系列高效液相色谱仪;美国AB公司API 4000三重四极杆串联质谱仪配电喷雾离子源;Dionex ASE 200型加速溶剂萃取仪(美国戴安公司);40 mL棕色玻璃瓶,带内衬有聚四氟乙烯膜的螺旋盖。
1.2 空白土壤样品、标准物质和主要试剂
空白土壤样品:采自北京郊区,不含待测目标物,阴干,混匀过40目筛。
土壤标准物质:ASA-1a(辽宁棕壤)、ASA-3a(四川紫色土)、ASA-5a(江西红壤)、ASA-6a(广东水稻土)、ASA-9(陕西黄绵土),均购自中国地质科学院地球物理地球化学勘查研究所。
18种氨基甲酸酯及其降解产物、三唑类农药单体标准品(100 mg/L,均购自中国计量科学研究院)。
内标物:甲萘威-D3、涕灭威-D3、戊唑醇-D6(100 mg/L,均购自德国Dr.E公司)。
替代物:氟康唑-D4(100 mg/L,德国Dr.E公司),4-溴-3, 5-二甲苯基-N-甲基氨基甲酸酯(BDMC,100 mg/L,百灵威化学有限公司)。
乙腈、甲醇、丙酮、二氯甲烷(HPLC级,百灵威化学有限公司)。
1.3 实验方法
1.3.1 加速溶剂萃取实验方法
准确称取5.00 g空白土壤样品和3.00 g硅藻土置于50 mL小烧杯中,搅拌均匀后装入22 mL ASE不锈钢萃取池(萃取池底部加纤维滤膜),加入目标化合物和替代物标准氟康唑-D4和BDMC,将萃取池置于加速溶剂萃取仪上用乙腈进行样品的提取,提取温度为70℃,静态提取时间5 min,循环次数3次,冲洗体积60%,氮气吹扫时间60 s。提取液经旋转蒸发浓缩和氮气吹干后,以甲醇-水(V:V=1:1) 定容至1 mL,0.22 μm滤膜过滤,待LC-MS/MS测定,内标法定量。
1.3.2 超声提取实验方法
准确称取5.00 g空白土壤样品于40 mL棕色玻璃瓶中,加入目标化合物和替代物标准氟康唑-D4和BDMC,涡旋混匀后加入20 mL提取溶剂乙腈。将玻璃瓶盖旋紧后置于超声波机中在室温下提取15 min后涡旋1 min,重复上述过程两次后离心15 min(转速3000 r/min),取上清液4 mL氮气吹干,以甲醇-水(V:V=1:1) 定容至1 mL,0.22 μm滤膜过滤,待LC-MS/MS测定,内标法定量。
1.3.3 QuEChERS提取实验方法
准确称取5.00 g空白土壤样品于40 mL棕色玻璃瓶中,加入目标化合物和替代物标准氟康唑-D4、BDMC和20 mL提取溶剂丙酮-二氯甲烷(V:V=1:1) 涡旋混匀。旋紧玻璃瓶盖后置于振荡器上以230 r/min振荡60 min后离心20 min(转速3000 r/min)。取上清液4 mL氮气吹干,以甲醇-水(V:V=1:1) 定容至1 mL,0.22 μm滤膜过滤,待LC-MS/MS测定,内标法定量[12]。
1.3.4 净化方法
分散固相萃取(d-SPE)净化:称取150 mg的N-丙基乙二胺(PSA)、200 mg石墨化碳黑(GCB)和500 mg无水硫酸镁于15 mL聚丙烯离心管中,取离心后的提取液上清液8 mL加入含上述吸附剂的离心管中涡旋振荡1 min后离心20 min(流速3000 r/min),取上清液4 mL,氮气吹干,以1 mL甲醇-水(V:V=1:1) 定容,待LC-MS/MS测定。
氨基SPE净化:将目标物的混合标准溶液加入2 mL甲醇-二氯甲烷(V:V=1:99) 中制成模拟加标提取液, 将其加到氨基固相萃取柱(使用前先用4 mL上述甲醇-二氯甲烷活化)中重力过柱;用2 mL甲醇-二氯甲烷(V:V=1:99) 洗涤盛放模拟提取液的离心管,并将洗涤液加入固相萃取柱,重复操作两次;收集各段洗脱液后氮吹,浓缩至干,以甲醇-水(V:V=1:1) 定容至1 mL,0.22 μm滤膜过滤,待LC-MS/MS测定。
1.3.5 液相色谱-质谱分析条件
色谱条件:Waters carbamate专用分析柱(4.6 mm×150 mm;5 μm),柱温30℃。流动相:A为水相,B为甲醇相;流速0.3 mL/min;进样量40 μL。
质谱条件:离子源为ESI(电喷雾);电离模式为正离子;电喷雾电压IS 5000 V;离子源温度400℃;碰撞气压力68.9 kPa;气帘气压力68.9 kPa;雾化气压力137.9 kPa;扫描方式为多反应选择离子监测方式,各监测离子对及其质谱参数见文献[12]。
2. 结果与讨论
2.1 ASE提取方法对检测结果的优化
温度、提取溶剂是ASE提取的重要分析条件,其中温度对热稳定性差的目标物有着更关键的影响。本文重点对ASE提取温度进行了优化,设定ASE提取温度为60~130℃,考察温度对基体加标样品回收率的影响。实验结果显示,三唑类杀菌剂基体加标回收率随温度升高而降低;氨基甲酸酯杀虫剂在70~100℃范围内萃取效率相对较高(43.8%~87.4%,甲硫威几乎没有回收),在温度超过100℃后绝大多数目标物回收率快速降低(38.0%~65.7%,甲硫威几乎没有回收),可能与高温下目标物发生了热降解有关。实验选择70℃作为ASE提取温度可达到较好的回收效果。
2.2 QuEChERS提取方法的优化
2.2.1 提取溶剂的优化
本文对提取溶剂进行优化,分别考察甲醇、乙腈、乙腈-甲醇(V:V=2:1)、丙酮、丙酮-二氯甲烷(V:V=3:1) 等多种提取溶剂对土壤中农药组分提取率的影响(图 1)。实验结果表明丙酮对所有目标物的提取回收率普遍偏低;甲醇、乙腈-甲醇对氨基甲酸酯类农药的回收率偏低;使用乙腈的基体加标回收率变化比较大(48.1%~125.9%),如涕灭威亚砜为48.1%,三羟基克百威、涕灭威砜、丙环唑、戊唑醇和己唑醇等超过120%;而丙酮-二氯甲烷的基体加标回收率保持在65.2%~113.9%之间,满足分析要求。因此,实验选择丙酮-二氯甲烷(V:V=3:1) 作为QuEChERS方法的提取溶剂。
![]() 图 1 QuEChERS提取溶剂对目标物回收率的影响Figure 1. Effect of the solvent of QuEChERS extraction on recoveries of target compounds
图 1 QuEChERS提取溶剂对目标物回收率的影响Figure 1. Effect of the solvent of QuEChERS extraction on recoveries of target compounds2.2.2 净化方法的优化
基质效应对液质联用的定量分析会产生较大的影响,在复杂基质的多农残检测中很有必要对其净化方法进行研究。将Anastassiades等[5]建立的d-SPE和目前应用广泛效果较好的氨基SPE小柱净化的回收率进行对比,对模拟的样品提取液进行d-SPE和SPE净化实验,考察净化效果。结果表明:d-SPE净化回收率在75.1%~108.7%之间,同SPE净化结果相当。d-SPE净化操作简单易于掌握,不需要额外消耗有机溶剂,成本与SPE相比降低了数倍,样品通量大,因此实验选择d-SPE对样品提取液进行净化。
2 3三种提取方法对比研究
2.3.1 三种提取方法的回收率对比
在上述优化的条件下分别采用ASE、UE和土壤QuEChERS方法对加标浓度为10 μg/kg的18种氨基甲酸酯和三唑类农药土壤样品进行了提取和LC-MS/MS测定,测定结果见表 1。总体来看,ASE提取目标物的回收率普遍较低,除甲萘威、涕灭威亚砜、三羟基克百威达到70%以上外,大多数目标物回收率不能满足分析要求,特别是甲硫威、涕灭威、三唑醇三种组分几乎没有回收。这可能与ASE属较高温压条件下萃取,造成热稳定性差的氨基甲酸酯、三唑类农药发生降解有关。UE的基体加标回收率除涕灭威为4.7%、涕灭威亚砜为137.8%之外,其他组分回收率为71.0%~100.0%。可见改进后的QuEChERS方法比另两种提取方法结果更优,除涕灭威回收率仍然偏低外(27.5%),其他组分回收率均为76.3%~121.0%,能够用于土壤中氨基甲酸酯类和三唑类农药的提取,推断与氨基甲酸酯类和三唑类农药既具有较强极性又具有一定脂溶性,易于从土壤转移到丙酮-二氯甲烷这种极性和亲酯较强的溶剂中有关。
表 1 三种提取方法加标回收率对比Table 1. Comparison of spiked recoveries for targets using different sample extraction methods目标物 基体加标回收率(%) ASE UE QuEChERS 克百威 64.9 86.2 100.7 残杀威 67.5 84.2 91.9 仲丁威 54.7 82.5 89.1 甲萘威 85.1 92.4 100.4 异丙威 52.6 77.1 83.0 速灭威 43.4 71.0 76.3 甲硫威 0.5 82.3 108.7 三羟基克百威 77.6 92.0 97.6 涕灭威 0.0 4.7 27.5 涕灭威砜 31.1 100.0 101.9 涕灭威亚砜 81.1 137.8 121.0 杀线威 60.7 84.1 93.2 灭多威 34.1 94.7 88.1 丙环唑 44.8 87.6 102.9 戊唑醇 56.0 92.6 92.4 烯唑醇 65.7 77.0 90.6 己唑醇 63.6 91.3 86.7 三唑醇 0.3 97.0 89.2 2.3.2 提取热效应对涕灭威、涕灭威亚砜回收率的影响
通过ASE、UE、QuEChERS三种提取方法对涕灭威、涕灭威亚砜对比研究发现,涕灭威稳定性非常差,极易受热转变成涕灭威亚砜。ASE属于加热提取,涕灭威受热分解并部分转化成涕灭威亚砜,而且涕灭威亚砜也存在部分热分解,回收率仅达到81.1%;UE虽然不加热,但它也存在一定的热效应,导致涕灭威回收率较低仅有4.7%,涕灭威亚砜则偏高达到137.8%;QuEChERS法采用振荡提取,热效应不显著,涕灭威回收率有较大提高(27.5%),但仍然存在降解,使得涕灭威亚砜回收率偏高至121.0%,但还在正常范围内(图 2)。因此,可以用涕灭威、涕灭威砜和涕灭威亚砜三者总量来表征涕灭威总污染水平。
![]() 图 2 提取方法对涕灭威、涕灭威亚砜检测结果的影响Figure 2. Effect of different extraction methods on spiked recoveries of aldicarb and aldicarb sulfoxide
图 2 提取方法对涕灭威、涕灭威亚砜检测结果的影响Figure 2. Effect of different extraction methods on spiked recoveries of aldicarb and aldicarb sulfoxide2.4 QuEChERS方法的技术指标和验证
2.4.1 QuEChERS方法的线性范围、检出限与精密度
配制不同浓度水平的目标物混合标准系列溶液,得到各目标物的线性范围、线性相关系数及回归方程;以空白土壤样品为基质进行低浓度水平加标实验,依据美国EPA方法计算各目标物的方法检出限,结果见表 2。在10 μg/kg的土壤添加浓度水平进行8次平行实验,相对标准偏差(RSD)在2.0%~16.8%之间(表 2)。
表 2 目标物的线性范围、回归方程、相关系数、检出限及精密度Table 2. Linear ranges, regression equation, correlation coefficients, detection limits and precision of target compounds目标物 线性范围(μg/L) 回归方程 相关系数(r2) 方法检出限(μg/kg) RSD (%) 克百威 0.1~100 y=0.9927x+0.0135 0.9999 0.010 5.4 残杀威 0.1~100 y=0.7924x-0.0095 0.9991 0.020 8.7 仲丁威 0.1~100 y=1.4403x-0.0659 0.9980 0.025 11.0 甲萘威 0.1~100 y=1.0581x-0.0001 0.9986 0.030 8.1 异丙威 0.1~100 y=0.9545x-0.0239 0.9996 0.020 9.7 速灭威 0.1~100 y=1.3277x-0.0528 0.9989 0.060 8.2 甲硫威 0.1~20 y=0.8937x+0.0317 0.9967 0.025 10.1 三羟基克百威 0.5~100 y=0.2795x-0.0284 0.9958 0.120 6.3 涕灭威 0.1~100 y=0.7502x-0.0321 0.9997 0.025 3.7 涕灭威砜 0.1~10 y=0.2450x+0.0056 0.9995 0.010 2.3 涕灭威亚砜 0.1~10 y=4.9287x+0.0418 0.9997 0.030 4.2 杀线威 0.5~20 y=1.1285x-0.0068 0.9987 0.075 5.3 灭多威 0.2~10 y=0.4174x+0.0133 0.9991 0.130 2.0 丙环唑 0.1~100 y=0.8749x-0.1068 0.9956 0.025 9.3 戊唑醇 0.1~100 y=1.2944x-0.0881 0.9981 0.015 7.9 烯唑醇 0.1~100 y=0.4856x-0.0701 0.9954 0.040 12.9 己唑醇 0.1~100 y=0.5812x-0.0481 0.9967 0.035 14.8 三唑醇 0.1~100 y=0.2806x-0.0021 0.9961 0.015 16.8 注:x表示目标物浓度/内标物浓度;y表示目标物峰面积/内标物峰面积。 2.4.2 不同类型土壤对QuEChERS方法提取结果的影响
选取辽宁棕壤、四川紫色土、江西红壤、广东水稻土、陕西黄绵土等5种不同类型的土壤标准样品进行加标回收实验(每种土壤类型均设平行样S1、S2),加标浓度10 μg/kg,实验结果见表 3。结果表明克百威、残杀威、异丙威、速灭威、三羟基克百威、涕灭威砜、涕灭威亚砜、杀线威、灭多威等组分的基体加标回收率为63.6%~114.7%,基本满足分析测试要求,其基体加标回收率与土壤类型相关性不大;而仲丁威、甲萘威、甲硫威、丙环唑、戊唑醇、烯唑醇、己唑醇、三唑醇等组分的基体加标回收率为13.2%~145.3%,显著与土壤类型相关,如广东水稻土中己唑醇的基体加标平均回收率仅为19.9%,而四川紫色土的基体加标平均回收率则达到74.0%;热稳定性差的涕灭威则在5种土壤类型中基体加标回收率均不理想,最高仅为21.3%。因此,在实际样品测试过程中要充分考虑样品基质对检测结果的影响。
表 3 不同类型土壤样品的加标回收率Table 3. Spiked recoveries of different soil types目标物 辽宁棕壤
加标回收率(%)四川紫色土
加标回收率(%)江西红壤
加标回收率(%)广东水稻土
加标回收率(%)陕西黄绵土
加标回收率(%)S1 S2 S1 S2 S1 S2 S1 S2 S1 S2 克百威 96.3 103.1 104.7 103.7 102.1 97.9 73.8 75.4 85.9 85.3 残杀威 85.6 88.9 84.3 96.1 88.2 90.2 73.9 77.1 83.7 75.8 仲丁威 83.5 90.3 90.9 78.2 96.8 104.7 55.0 57.6 88.5 84.4 甲萘威 87.3 83.3 91.0 88.2 100.5 85.1 57.5 70.6 86.4 86.4 异丙威 73.7 85.6 89.4 82.6 92.4 99.6 63.6 66.9 87.3 81.4 速灭威 81.5 78.0 78.0 75.6 77.5 79.0 66.7 77.5 78.0 84.0 甲硫威 82.8 78.1 81.3 72.3 95.3 105.9 48.4 55.1 94.9 88.7 三羟基克百威 107.8 114.7 100.0 108.1 96.6 109.4 88.1 79.4 79.1 88.8 涕灭威 7.5 8.2 8.3 8.2 14.1 14.5 9.4 9.7 20.1 21.3 涕灭威砜 91.8 85.7 86.8 89.0 87.9 93.4 86.8 80.2 81.3 88.5 涕灭威亚砜 75.1 71.4 86.8 83.1 64.6 65.8 68.5 73.5 98.2 94.7 杀线威 68.9 65.8 68.5 69.1 71.9 85.8 84.3 79.9 90.6 93.8 灭多威 79.5 71.6 81.9 79.2 63.8 70.9 65.0 66.7 77.8 81.7 丙环唑 63.3 71.3 93.9 84.3 64.2 52.2 50.6 47.8 77.1 64.6 戊唑醇 55.8 75.0 97.0 96.1 40.1 13.2 30.4 29.4 72.8 63.0 烯唑醇 59.7 71.9 70.6 109.3 110.9 89.1 31.8 33.2 64.0 62.2 己唑醇 31.8 35.8 74.9 73.1 30.8 28.5 21.6 18.2 70.3 50.1 三唑醇 71.8 83.9 145.3 126.3 45.4 61.4 34.2 33.5 85.4 67.8 3. 结论
本文在对比ASE、UE和QuEChERS三种提取方法的基础上,研究优化了QuEChERS法提取土壤样品中18种痕量氨基甲酸酯和三唑类农药的方法。优化后的QuEChERS方法与ASE、UE相比提取效果更理想,17种组分回收率在76.3%~121.0%之间,涕灭威回收率也有明显提高,涕灭威亚砜回收率降至正常水平。研究还表明涕灭威回收率偏低、涕灭威亚砜回收率偏高与提取方法的热效应有关;部分组分如仲丁威、甲萘威的回收状况与土壤类型相关。总之,尽管QuEChERS方法在土壤的应用中受到一定限制,但本文研究结果表明优化后的QuEChERS方法还是可以应用于土壤中一些极性较强、热稳定较差的有机污染物的快速分析。
-
![]()
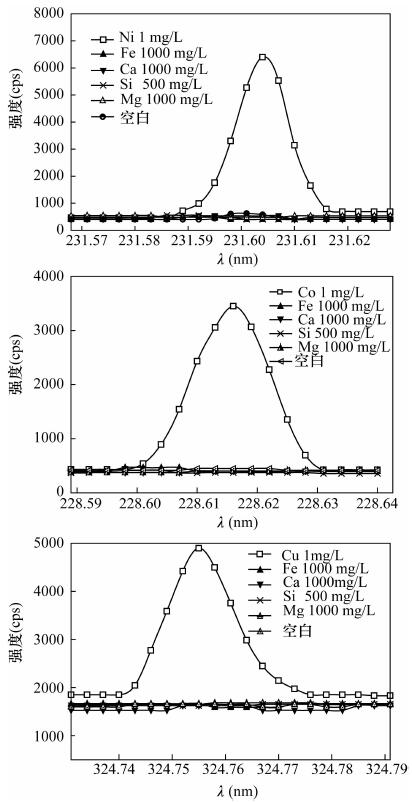
图 1 Ni 231.604 nm、Co 228.616 nm和Cu 324.754 nm波长处光谱干扰描迹图
Figure 1. The interference curves of Ni 231.604 nm, Co 228.616 nm and Cu 324.754 nm
表 1 红土镍矿主要成分
Table 1 The main components of nickel laterite
组分 含量(%) 组分 含量(%) Fe 7~56 TiO 0.004~0.09 SiO2 4~50 Cr2O3 0.8~5 MgO 0.5~35 Ni 0.3~3 Al2O3 1~9 Cu 0~0.01 CaO 0.03~3 P 0~0.03 MnO 0.1~0.4 Co 0.02~0.2  下载: 导出CSV
下载: 导出CSV
表 2 铁基体对待测元素测定的影响
Table 2 Effect of iron matrix on element determination
元素 a1/a2 b1-b2 元素 a1/a2 b1-b2 Ca 0.926 3.03 As 0.929 -0.105 Mg 0.952 -0.413 Zn 0.811 3.356 Al 0.984 -3.16 Cr 0.944 -0.682 Mn 0.864 -5.82 V 0.902 0.424 Cu 0.947 1.08 Ti 0.946 0.241 Pb 0.861 0.642 Co 0.859 -0.459 Ni 0.802 -1.54 Cd 0.887 -1.19 注:a1为加基体标准系列标准曲线的斜率,a2为纯标准系列标准曲线的斜率;b1为加基体标准系列标准曲线的截距,b2为纯标准系列标准曲线的截距。
下载: 导出CSV
表 3 红土镍矿样品前处理方法和分析测定技术
Table 3 Sample preparation methods and analytical techniques for nickel laterite
测定元素 前处理方法 测定技术 测定干扰消除 文献 Ca,Mg 酸溶法:0.1 g试样,以20 mL盐酸-10 mL硝酸-5 mL氢氟酸-5 mL高氯酸分解。以盐酸溶解盐类 FAAS Ca 422.7 nm、Mg 285.2 nm。测镁时试液中添加0.002%氯化锶;测钙时试液中添加0.001%氯化锶 [14] Cd 酸溶法:0.2 g试样,以15 mL盐酸-5 mL硝酸-5 mL氢氟酸-3 mL高氯酸分解。以硝酸溶解盐类 FAAS Cd 228.8 nm [14] Co 酸溶法:0.2 g试样,以20 mL盐酸-10 mL硝酸-5 mL氢氟酸-5 mL高氯酸分解。以盐酸溶解盐类 FAAS Co 240.7 nm [14] Co,Ni 酸溶法:0.1~0.5 g试样,以5 ~10 mL氢氟酸,5 ~10 mL现配王水,0.5 ~1 mL高氯酸分解。以盐酸溶解盐类 FAAS Co 240.7 nm、Ni 232.0 nm或352.3 nm [15] Cr 酸溶法:0.2 g试样,以2 mL磷酸-2 mL硫酸-5 mL硝酸-2 mL高氯酸分解 FAAS Cr 357.9 nm;采用空气-乙炔贫焰;铁基体匹配标液及空白;加入硫酸钠-氯化铵作为干扰抑制剂 [24] Cr,Cu,Zn 酸溶法:0.5 g试样,以15 mL盐酸-5 mL硝酸-5 mL氢氟酸-1 mL高氯酸分解。以硝酸溶解盐类。
碱熔法:0.5 g试样,以1.3 g碳酸钠-2.7 g过氧化钠800℃熔融,20 mL盐酸浸提。采用刚玉坩埚。
酸溶-焦硫酸钾熔融残渣法:0.5 g试样,以30 mL盐酸-10 mL硝酸分解,残渣用5~6 g焦硫酸钾700℃熔融,热水浸提。采用铂坩埚FAAS Cr 357.9 nm、Cu 324.8 nm、Zn 213.9 nm [7, 23] Cu 酸溶法:0.5 g试样,以15 mL盐酸-5 mL硝酸-5 mL氢氟酸-1 mL高氯酸分解。以硝酸溶解盐类 FAAS Cu 324.8 nm [14] Mn 酸溶法:0.1~0.2 g试样,以10 mL盐酸-5 mL硝酸-5 mL氢氟酸-2 mL高氯酸分解。以盐酸溶解盐类 FAAS Mn 279.5 nm [14] Ni 酸溶法:0.1 g试样,以6 mL盐酸-2 mL硝酸-2 mL氢氟酸-1 mL高氯酸分解。以盐酸溶解盐类 FAAS Ni 232.0 nm [14] Ni 酸溶法:0.1 g试样,以15 mL盐酸-5 mL硝酸分解。以盐酸溶解盐类。
碱熔法:0.1 g试样,以1 g过氧化钠600℃熔融,15 mL浓盐酸洗出坩埚FAAS Ni 232.0 nm [30] Pb 酸溶法:0.1~0.5 g试样,以15 mL盐酸-5 mL硝酸-5 mL氢氟酸-2 mL高氯酸分解。以硝酸溶解盐类 FAAS Pb 283.3 nm [14] Zn 酸溶法:0.1 g试样,以6 mL盐酸-2 mL硝酸-2 mL氢氟酸-0.5 mL高氯酸分解。以盐酸溶解盐类。
碱熔法:0.1 g试样,以4 g过氧化钠750℃熔融,20 mL 25%盐酸和1滴双氧水浸提。采用刚玉坩埚FAAS Zn 213.9 nm对于锌量小于0.05%,且铁量大于30%的红土镍矿,采用铁基体匹配标准溶液及空白 [22] Zn 酸溶法:0.1 g试样,以6 mL盐酸-2 mL硝酸-2 mL氢氟酸-0.5 mL高氯酸分解。以盐酸溶解盐类 FAAS Zn 213.9 nm [14] Hg 酸溶法:0.1~0.5 g试样,以10 mL盐酸-硝酸-水(1:3:4) 分解。加入重铬酸钾稳定。用氯化亚锡还原汞离子 冷原子AAS Hg 253.7 nm [14] Al,Cr,Fe,Mg,
Mn,Ni,Si压片法或熔融玻璃法制样 EDXRF Al 1.49 keV,Cr 5.42 keV,Fe 6.4 keV,Mg 1.42 keV,Mn 5.9 keV,Ni 7.48 keV,Si 1.74 keV [14] Ca,Cr,Fe,Mn,
Ni,Ti,Zn无需制样,直接将试样装入样品杯上机测试 便携式EDXRF - [10] Al,Ca,Cu,Fe,Mg,
Mn,Ni,P,Si,Ti熔融制样,试样与四硼酸锂-偏硼酸锂(12:22) 混合熔剂以1:10比例混匀 WDXRF 熔融制样消除样品不均匀性及粒度效应。利用可变理论α影响系数法校正元素间的吸收增强效应 [11] Al,Ca,Cr,Co,Fe,
Mg,Mn,Ni,P,Si,Ti熔融制样,试样与四硼酸锂-偏硼酸锂(67:33) 混合熔剂以1:10比例混匀 WDXRF 根据实际情况选择合适的校准方程,如理论α影响系数法、基本参数法、经验α影响系数法等。测定Co时,须校正Fe Kβ1对Co Kα的谱线重叠影响 [15] Al,Ca,Cr,Co,Fe,
Mg,Mn,Ni,P,Si,Ti熔融制样,试样与四硼酸锂-偏硼酸锂(12:22) 混合熔剂以1:10比例混匀 WDXRF 根据实际情况选择合适的校准方程。测定Co时,须校正Fe Kβ1对Co Kα谱线重叠影响 [14] Al,Ca,Cr,Co,Fe,
K,Mg,Mn,Ni,P,
S,Si,Ti,V熔融制样,试样与四硼酸锂-偏硼酸锂(67:33) 混合熔剂以1:10比例混匀 WDXRF 在样品中加入少量氧化剂硝酸锂抑制S元素在高温下的损失。采用理论α影响系数法校正元素间的吸收增强效应 [12] Al,Ca,Cr,Co,F
e,Mg,Mn,Ni,Si压片法或熔融玻璃制样熔融制样:试样与四硼酸锂-偏硼酸锂(67:33) 混合熔剂以1:20比例混匀,1150℃熔融 WDXRF 针对压片制得的样品采用康普顿散射线作内标和经验系数法校正基体效应。针对熔融制得的熔片采用理论α影响系数法校正元素间的吸收增强效应 [13] Co,Cu,Ni 碱熔法:0.2 g试样,以0.9 g碳酸钠-四硼酸钠(2:1) 混合熔剂1000℃熔融,以20 mL 50%盐酸浸提。采用铂金坩埚 ICP-AES Co 228.616nm、Ni 231.604 nm使用Y 224.306 nm作为内标线。Cu 324.754 nm使用Y 371.030 nm作为内标线 [26] Ca,Cd,Co,Cu,
Mg,Mn,Ni,Pb,Zn酸溶法:0.1 g试样,以5 mL盐酸-15 mL硝酸-3 mL氢氟酸分解。以盐酸溶解盐类 ICP-AES Cd 228.802 nm,Co 237.862 nm,Cu 324.754 nm,Mg 285.213 nm,Mn 257.610 nm,Ni 221.647 nm,Pb 220.353 nm,Zn 213.856 nm,Ca 393.366 nm [27] Al,Ca,Cr,Co,Mg,
Mn,Ni,Si,Ti碱熔法:0.5 g试样,以0.8 g碳酸钠-0.4 g硼酸950℃熔融,以盐酸浸提。采用铂金坩埚 ICP-AES Si 251.611 nm,Ca 315.887 nm,Mg 279.553 nm,Al 308.215 nm,Mn 257.610 nm,Ti 323.452 nm,Cr 267.716 nm,Ni 216.556 nm,Co 228.616 nm。采用铁基体匹配标准溶液 [28] Al,Co,Mg,Ni 酸溶法:0.2 g试样,以(5~10 mL)氢氟酸-(5~10 mL)现配王水-5 mL高氯酸分解。以盐酸溶解盐类 ICP-AES Ni 231.604 nm,Mg 383.829 nm,Al 308.22 nm,Co 228.616 nm。采用铁基体匹配标准溶液 [6] Ca,Co,Cu,Mg,
Mn,Ni,P,Zn酸溶法:0.2 g试样,以20 mL盐酸-10 mL硝酸-10 mL氢氟酸-5 mL高氯酸分解。以盐酸溶解盐类 ICP-AES Ca 317.933 nm,Co 228.616 nm,Cu 327.396 nm,Mg 280.271 nm,Mn 257.610 nm,Ni 231.604 nm,P 178.287 nm,Zn 206.200 nm [14] Al,Co,Mg,Ni,P 酸溶法:0.2 g试样,以6 mL盐酸-4 mL硝酸-5 mL氢氟酸-1.5 mL高氯酸分解。以盐酸溶解盐类 ICP-AES Ni 231.604 nm,Mg 383.829 nm,Al 308.22 nm,Co 228.616 nm,P 213.618 nm。采用铁基体匹配标准溶液 [15] Al,Co,Mg,Ni,Fe 酸溶法:0.1 g试样,以10 mL王水-5 mL氢氟酸-3 mL高氯酸分解。以盐酸溶解盐类 ICP-AES Ni 231.604 nm,Mg 280.270 nm,Al 394.401 nm,Co 228.616 nm,Fe 238.204 nm [33] Ni 微波消解:0.1 g试样,以6 mL盐酸-2 mL硝酸-1 mL氢氟酸,之后以10 mL 5%硼酸中和氢氟酸 ICP-AES Ni 231.604 nm,使用Y 371.030 nm作为内标线 [25] Ni 酸溶法:0.1 g试样,以15 mL盐酸-5 mL硝酸-2 mL氢氟酸-2 mL高氯酸分解。以硝酸溶解盐类 UV-vis 酒石酸钾钠掩蔽试液中的共存干扰金属离子。以不加丁二酮肟显色剂的试液作参比。460 nm测定 [21] Ni 酸溶法:0.1 g试样,以15 mL盐酸-5 mL硝酸-2 mL氢氟酸-2 mL高氯酸分解。以硝酸溶解盐类 UV-vis 酒石酸钾钠掩蔽试液中的共存干扰金属离子。在±5%误差内,2.0 μg/mL Ni不受共存离子的干扰。460 nm检测 [31] Ni 酸溶法:1.0 g试样,以15 mL盐酸-5 mL硝酸-5 mL氢氟酸-5 mL高氯酸分解。以水溶解盐类 UV-vis 酒石酸钾钠掩蔽试液中的共存干扰金属离子。530 nm检测 [32] Ni 酸溶法:0.1 g试样,以15 mL盐酸-5 mL硝酸-2 mL氢氟酸-2 mL高氯酸分解。以硝酸溶解盐类 UV-vis 酒石酸钾钠掩蔽试液中的共存干扰金属离子。以试剂空白作参比。460 nm测定 [14] P 酸溶法:0.2~0.4 g试样,以10 mL盐酸-10 mL硝酸-(5~10 m)L氢氟酸-5 mL高氯酸分解 UV-vis 盐酸-氢溴酸消除As干扰。690 nm测定 [14] As,Bi,Sb 酸溶法:0.2 g试样,以12 mL王水,沸水浴加热 AFS 在硫脲-抗坏血酸溶液存在下,共存元素对待测元素不存在干扰 [18] As 酸溶法:0.2 g试样,以10 mL硝酸-5 mL 0.1%氟化氢铵-2 mL硫酸分解。以盐酸溶解盐类 AFS 在硫脲-抗坏血酸溶液存在下,共存元素对待测元素不存在干扰 [19] As,Bi,Sb 酸溶法:0.2 g试样,以10 mL硝酸-5 mL 0.1%氟化氢铵-2 mL硫酸分解。以盐酸溶解盐类 AFS 在硫脲-抗坏血酸溶液存在下,共存元素对待测元素不存在干扰 [14] Sc,Cd 酸溶法:0.1 g试样,以10 mL盐酸-5 mL硝酸-5 mL氢氟酸-2 mL高氯酸分解。以硝酸溶解盐类 ICP-MS - [14] F,Cl 水蒸气蒸馏法:0.5 g试样,以33%的硫酸分解,目标元素随水蒸气逸出,经10 mL 0.2 mol/L氢氧化钠吸收 离子色谱法 - [14] Al 碱熔法:0.2 g试样,以5 g氢氧化钠650℃熔融,以沸水浸提。采用银坩埚 EDTA滴定法 - [14] 全铁 碱熔法:0.2~0.4 g试样,以3~4 g过氧化钠800℃熔融,以盐酸浸提。采用刚玉坩埚 重铬酸钾
滴定法30 mg Mg、Si, 10 mg Ni、Cr2O3不干扰Fe的测定 [34] Mg 酸溶法:0.2 g试样,以10 mL盐酸-10 mL硝酸-(5~8 mL)氢氟酸-3 mL高氯酸分解。以盐酸溶解盐类 EDTA
滴定法以六次甲基四胺、二乙胺基二硫代甲酸钠沉淀分离大量共存金属离子 [14] SiO2 碱熔法:0.1~0.2 g试样,以4 g氢氧化钾650℃熔融,以沸水浸提。采用镍坩埚 氟硅酸钾
容量法选用氢氧化钾作熔剂,硝酸介质,缩短沉淀放置时间消除40 mg Al的干扰 [20] C,S 高频燃烧熔融:0.1 g试样,以1.0 g纯铁-(1.0~1.5 g)钨锡(9:1) 混合助溶剂熔样 红外吸收
光谱法- [14] S 管式电阻炉加热熔融:0.2 g试样,以0.2 g纯铁,约2 g线状氧化铜熔样 红外吸收
光谱法- [35] 注:EDXRF为能量色散X射线荧光光谱法;WDXRF为波长色散X射线荧光光谱法。
下载: 导出CSV
-
-
期刊类型引用(15)
1. 刘毅,李裴彤,郭守武,罗威,张利锋. V_2AlC粉体的形貌调控及其吸波性能研究. 陕西科技大学学报. 2024(01): 118-124 .  百度学术
百度学术
2. 何妮,李尚颖,梁绍暹,周轩平,武振晓,赵昊,汪洋,程宏飞. 铵伊利石的矿物学特征及热稳定性. 地球科学与环境学报. 2024(02): 229-239 . 百度学术
3. 张敏,龚沂,陈赟,赵天伟,屈叶飞. 不同样品中游离二氧化硅分析研究进展. 分析仪器. 2024(04): 1-5 . 百度学术
4. 张利锋,王凯,王晓飞,刘毅,郭守武. 硅藻土衍生硅碳负极材料的制备及其储锂性能研究. 陕西科技大学学报. 2024(05): 119-125 . 百度学术
5. 张敏,龚沂,陈赟,赵天伟,屈叶飞. 电感耦合等离子体发射光谱法测定石灰岩中游离二氧化硅. 化学分析计量. 2024(10): 45-50 . 百度学术
6. 乔柱,郑建明,宋见峰,朱晓东,封亚辉. Si内标-X射线衍射法定量测定铁矿中Fe_3O_4、Fe_2O_3和SiO_2. 中国无机分析化学. 2023(05): 463-468 . 百度学术
7. 周君龙,陈宜浩,李全忠,赵泉,唐志锟,宋武元. X射线衍射Rietveld全谱拟合法定量检测滑石混合矿物中滑石含量. 冶金分析. 2023(04): 18-23 . 百度学术
8. 李磊,张青,范慧,阳珊,遇祯. 土壤中高岭石的X射线衍射全谱拟合定量分析方法研究. 安徽地质. 2023(03): 282-287 . 百度学术
9. 谭杰,周克明,曾理,邹春艳,严玉霞,孔令明,袁小玲. 火山岩晶质及非晶质矿物组分定量分析方法——以四川盆地西部二叠系火山岩为例. 天然气工业. 2022(05): 24-33 . 百度学术
10. 张悦,江荣,张磊成,陈西辉,高希光,孙志刚,宋迎东. 直接烧结6H-SiC氧化机理的实验研究与分子动力学模拟. 航空材料学报. 2022(04): 28-38 . 百度学术
11. 王炳监,齐道正,陈登. 不同硫酸盐环境下铝酸三钙的劣化过程及机制. 无机盐工业. 2022(08): 119-124+150 . 百度学术
12. 唐梦奇,吕春秋,陈璐,冯均利,萧达辉. X射线衍射标准曲线法测定氧化锌富集物中游离态氧化锌. 冶金分析. 2021(07): 23-27 . 百度学术
13. 邵丹. 锂电池石墨负极材料纯度检测方法研究. 化工设计通讯. 2019(03): 147+151 . 百度学术
14. 冉敬,杜谷,王凤玉. X射线衍射全谱拟合法快速分析长石矿物含量. 岩矿测试. 2017(05): 489-494+449 . 本站查看
15. 刘贵民,杨忠须,张一帆,闫涛,魏敏. 基于正交试验法的超音速等离子喷涂钼涂层组织及性能研究. 兵工学报. 2016(08): 1489-1496 . 百度学术
其他类型引用(9)


计量
- 文章访问数: 1575
- HTML全文浏览量: 268
- PDF下载量: 46
- 被引次数: 24
 京公网安备 11010202008159号
京公网安备 11010202008159号